
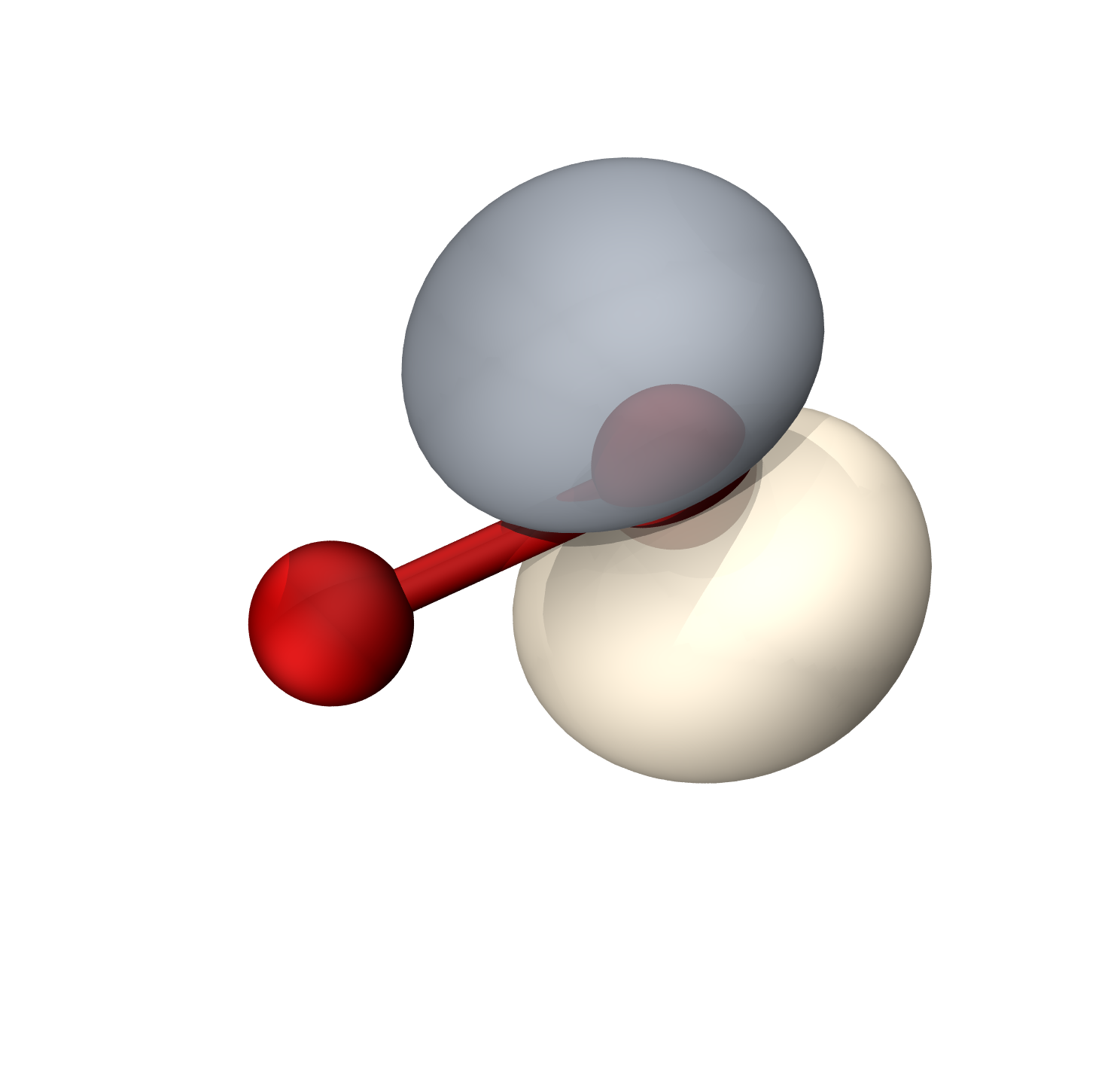
O2
分子轨道理论的处理
对于O2,可以使用CAS(2,2)(两个电子,两个π*轨道)描述
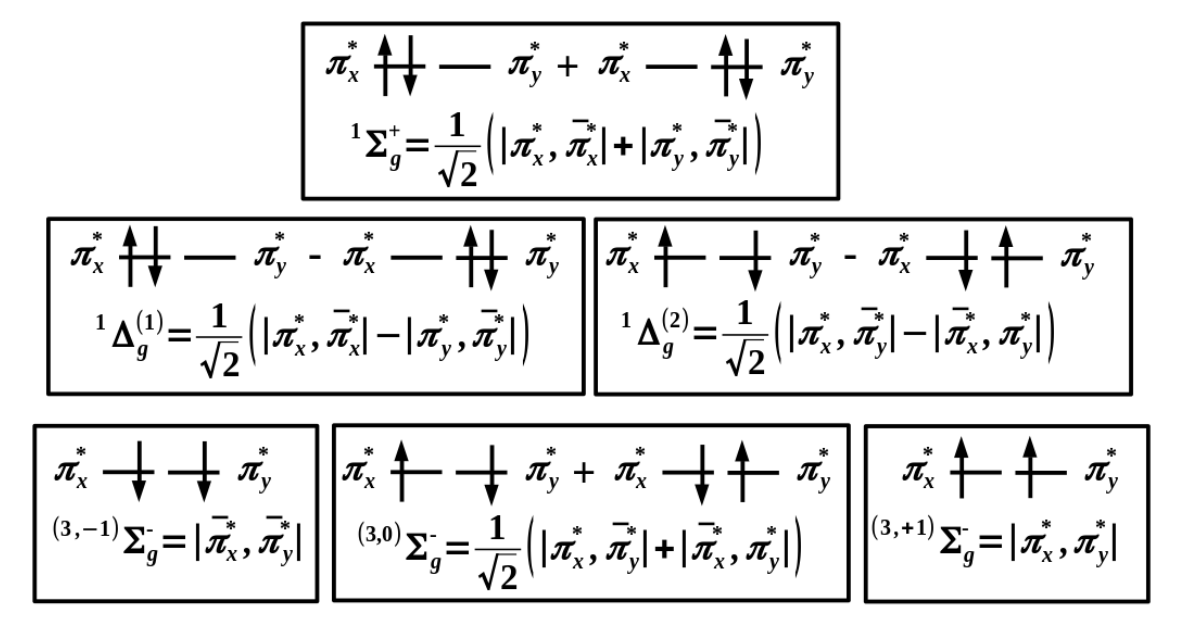 注意大多数教材对于单线态氧的描述并不正确
注意大多数教材对于单线态氧的描述并不正确
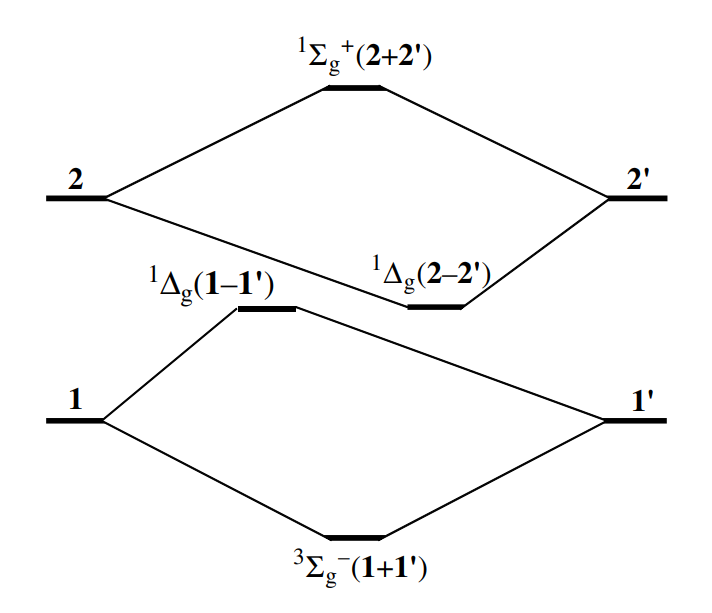
最下面是三个能量最低的三重态
中间是单重态: 和(自旋匹配组态)
能量最高的单重态为:
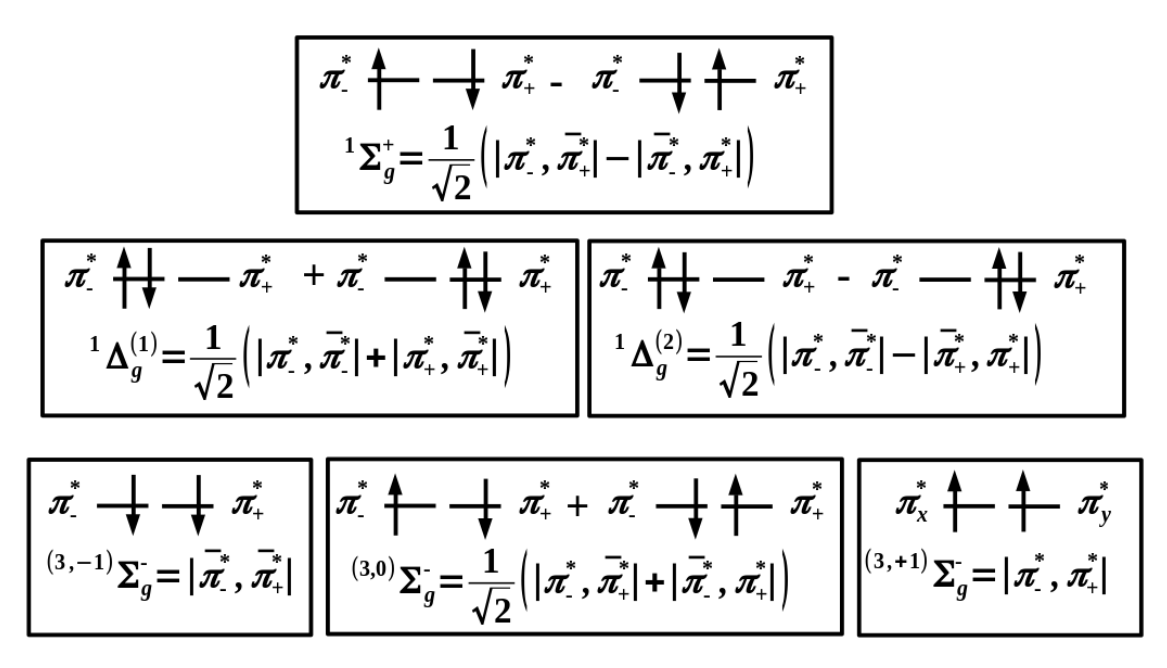
或者用复数轨道表示为:
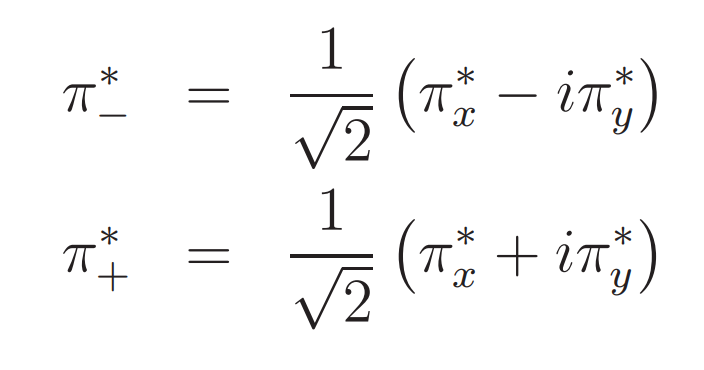
价键理论的处理
价键理论中最简单的情况,两种VB结构:
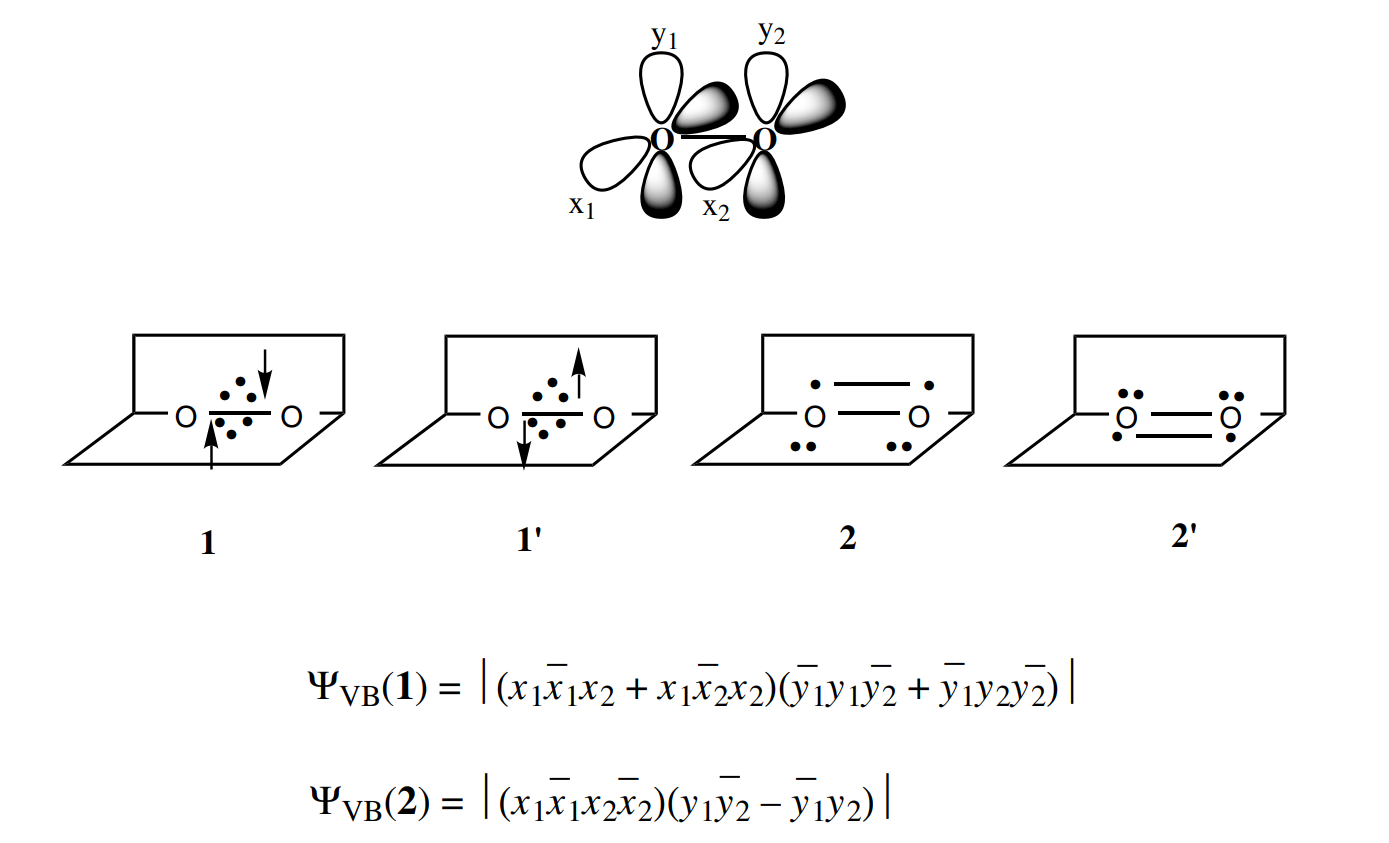
组合后:
计算
单重态
σ依旧保持离域,str=full生成所有结构
O2
$CTRL
str=full NAO=4 NAE=6 ISCF=5 IPRINT=3
ORBTYP=HAO FRGTYP=SAO
INT=LIBCINT BASIS=def2-TZVP
$END
$FRAG
2*1 1*4
SPZDXXDYYDZZ 1 2
PXDXZ 1
PXDXZ 2
PYDYZ 1
PYDYZ 2
$END
$ORB
1*9
1
1
1
1
1
2 # px
3
4 # py
5
$END
$GEO
O 0 0 0
O 0 0 1.21
$END能量: -149.64745
第一激发态能量也是-149.64745,符合二重简并的情况
查看系数可以发现这里算出来的是图中的(2-2’),并且还混入了1阶离子结构(7-10)
1 -0.58621751 ****** 1:5 6 6 7 7 8 9
2 0.00000000 ****** 1:5 6 6 8 8 7 9
3 0.00000000 ****** 1:5 6 6 9 9 7 8
4 0.00000000 ****** 1:5 7 7 8 8 6 9
5 0.00000000 ****** 1:5 7 7 9 9 6 8
6 0.58706331 ****** 1:5 8 8 9 9 6 7
7 0.14643850 ****** 1:5 6 6 7 7 8 8
8 0.14643846 ****** 1:5 6 6 7 7 9 9
9 -0.14660147 ****** 1:5 6 6 8 8 9 9
10 -0.14660154 ****** 1:5 7 7 8 8 9 9nstate=1
输入文件加入nstate=1计算激发态
系数如下
1 0.00000000 ****** 1:5 6 6 7 7 8 9
2 0.20720853 ****** 1:5 6 6 8 8 7 9
3 0.57126443 ****** 1:5 6 6 9 9 7 8
4 0.57126801 ****** 1:5 7 7 8 8 6 9
5 0.20720863 ****** 1:5 7 7 9 9 6 8
6 0.00000000 ****** 1:5 8 8 9 9 6 7
7 0.00000000 ****** 1:5 6 6 7 7 8 8
8 0.00000000 ****** 1:5 6 6 7 7 9 9
9 0.00000000 ****** 1:5 6 6 8 8 9 9
10 0.00000000 ****** 1:5 7 7 8 8 9 9nstate=2
能量: -149.62019
对应于图中(2+2’),混入了1阶离子结构(7-10)
1 -0.60926183 ****** 1:5 6 6 7 7 8 9
2 0.00000000 ****** 1:5 6 6 8 8 7 9
3 0.00000000 ****** 1:5 6 6 9 9 7 8
4 0.00000000 ****** 1:5 7 7 8 8 6 9
5 0.00000000 ****** 1:5 7 7 9 9 6 8
6 -0.60924638 ****** 1:5 8 8 9 9 6 7
7 0.12386817 ****** 1:5 6 6 7 7 8 8
8 0.12386791 ****** 1:5 6 6 7 7 9 9
9 0.12385947 ****** 1:5 6 6 8 8 9 9
10 0.12385942 ****** 1:5 7 7 8 8 9 9三重态
添加nmul=3计算三重态
能量: -149.67798
系数:
1 0.00000000 ****** 1:5 6 6 7 7 8 9
2 -0.24220602 ****** 1:5 6 6 8 8 7 9
3 -0.54551287 ****** 1:5 6 6 9 9 7 8
4 -0.54551280 ****** 1:5 7 7 8 8 6 9
5 -0.24211241 ****** 1:5 7 7 9 9 6 8
6 -0.00000000 ****** 1:5 8 8 9 9 6 7参考文献:
- Practical Treatment of Singlet Oxygen with Density-Functional Theory and the Multiplet-Sum Method
- A Chemist’s Guide to Valence Bond Theory
F2
第二周期双原子分子中有4个是强静态相关/RHF不稳定,F2是其中之一(其余三个是Be2,B2,C2)
F2 VBSCF with 3 structures
$CTRL
STR=FULL NAO=2 NAE=2 ISCF=5 IPRINT=3
ORBTYP=HAO FRGTYP=SAO
INT=LIBCINT BASIS=CC-PVTZ
$END
$FRAG
1*6
SPZDXXDYYDZZ 1
SPZDXXDYYDZZ 2
PXDXZ 1
PXDXZ 2
PYDYZ 1
PYDYZ 2
$END
$ORB
1*10
1
2
1
2
3
4
5
6
1
2
$END
$GEO
F 0.0 0.0 0.0
F 0.0 0.0 1.4
$ENDCharge-Shift Bonding
只有共价结构的F2是解离的,引入离子结构后才有F2成键势能面极小点
F2这种不寻常的键合被称为Charge-Shift Bonding
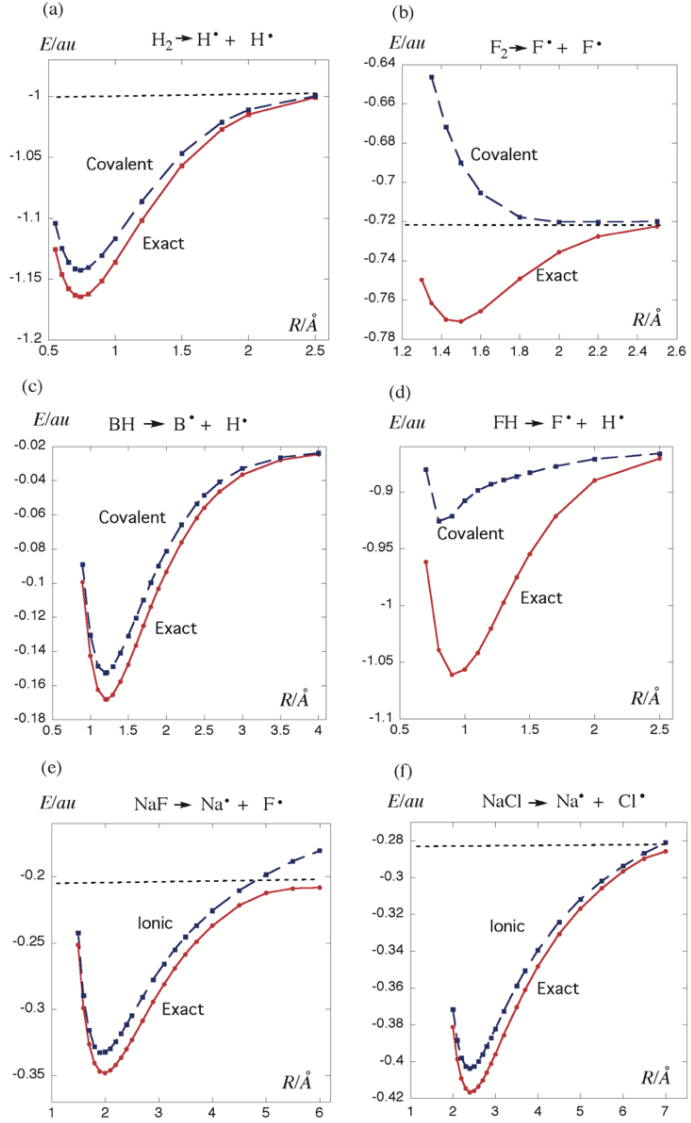
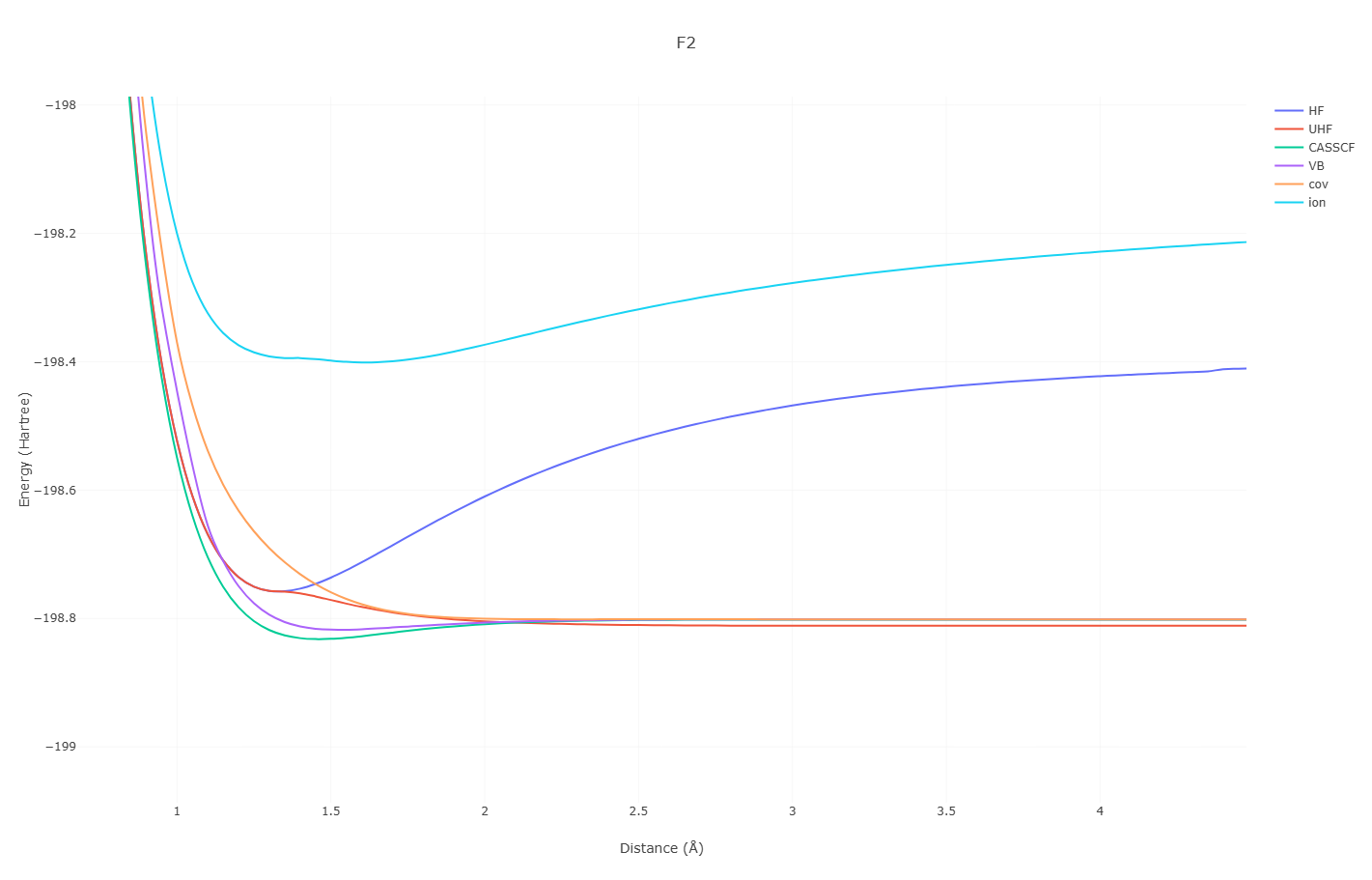
相关文献:
- Charge-Shift Bonding: A New and Unique Form of Bonding
- Covalent Bonding and Charge Shift Bonds: Comment on “The Carbon–Nitrogen Bonds in Ammonium Compounds Are Charge Shift Bonds”
BOVB
guess=read读取之前计算的轨道,添加BOVB关键词,ISCF改为1
F2 VBSCF with 3 structures
$CTRL
STR=FULL NAO=2 NAE=2 ISCF=1 IPRINT=3
BOVB guess=read
INT=LIBCINT BASIS=CC-PVTZ
$END
$FRAG
1*6
SPZDXXDYYDZZ 1
SPZDXXDYYDZZ 2
PXDXZ 1
PXDXZ 2
PYDYZ 1
PYDYZ 2
$END
$ORB
1*10
1
2
1
2
3
4
5
6
1
2
$END
$gus
13 13 13 13 5 5 5 5 13 13
# ORBITAL 1 NAO = 13
0.9712363790 1 -0.0039467143 2 0.0477867799 3 0.0013965990 4
0.0004229095 7 -0.0009534061 10 -0.0004443900 13 0.0023100215 14
0.0023103042 17 0.0020316376 19 0.0042243027 20 0.0042249670 23
0.0039211192 25
# ORBITAL 2 NAO = 13
0.9712174224 36 -0.0030546148 37 0.0480562273 38 0.0019225209 39
-0.0003610981 42 0.0010246535 45 0.0005038309 48 0.0023158300 49
0.0023117556 52 0.0020305383 54 0.0042379535 55 0.0042410917 58
0.0039419970 60
# ORBITAL 3 NAO = 13
-0.0153109841 1 0.5732512933 2 0.1777876180 3 0.3401362453 4
-0.0434593633 7 -0.0427231963 10 -0.0450025476 13 0.0033808736 14
0.0033798078 17 0.0017738680 19 0.0118949626 20 0.0118943278 23
0.0119190556 25
# ORBITAL 4 NAO = 13
-0.0168072743 36 0.5732576950 37 0.1777155826 38 0.3401329646 39
0.0434550684 42 0.0427190015 45 0.0449926489 48 0.0033761111 49
0.0033786322 52 0.0017710387 54 0.0118868786 55 0.0118868119 58
0.0119139175 60
# ORBITAL 5 NAO = 5
-0.3965889118 5 -0.4891751437 8 -0.3333067471 11 -0.0015485023 16
-0.0091428966 22
# ORBITAL 6 NAO = 5
-0.3965888567 40 -0.4891765077 43 -0.3333050528 46 0.0015475158 51
0.0091424354 57
# ORBITAL 7 NAO = 5
-0.3965889321 6 -0.4891751477 9 -0.3333067436 12 -0.0015465238 18
-0.0091424657 24
# ORBITAL 8 NAO = 5
-0.3965889167 41 -0.4891764943 44 -0.3333050219 47 0.0015462715 53
0.0091421766 59
# ORBITAL 9 NAO = 13
0.0007015120 1 -0.1830351617 2 -0.0732626576 3 0.0302573450 4
-0.3870316763 7 -0.5149651899 10 -0.2557597580 13 0.0001525648 14
0.0001525346 17 -0.0138751181 19 -0.0120108246 20 -0.0120108675 23
-0.0708024528 25
# ORBITAL 10 NAO = 13
0.0006399832 36 -0.1830304774 37 -0.0732655651 38 0.0302629387 39
0.3870318606 42 0.5149677701 45 0.2557604213 48 0.0001504536 49
0.0001505980 52 -0.0138758993 54 -0.0120087914 55 -0.0120087521 58
-0.0708013268 60
$END
$GEO
F 0.0 0.0 0.0
F 0.0 0.0 1.4
$END