
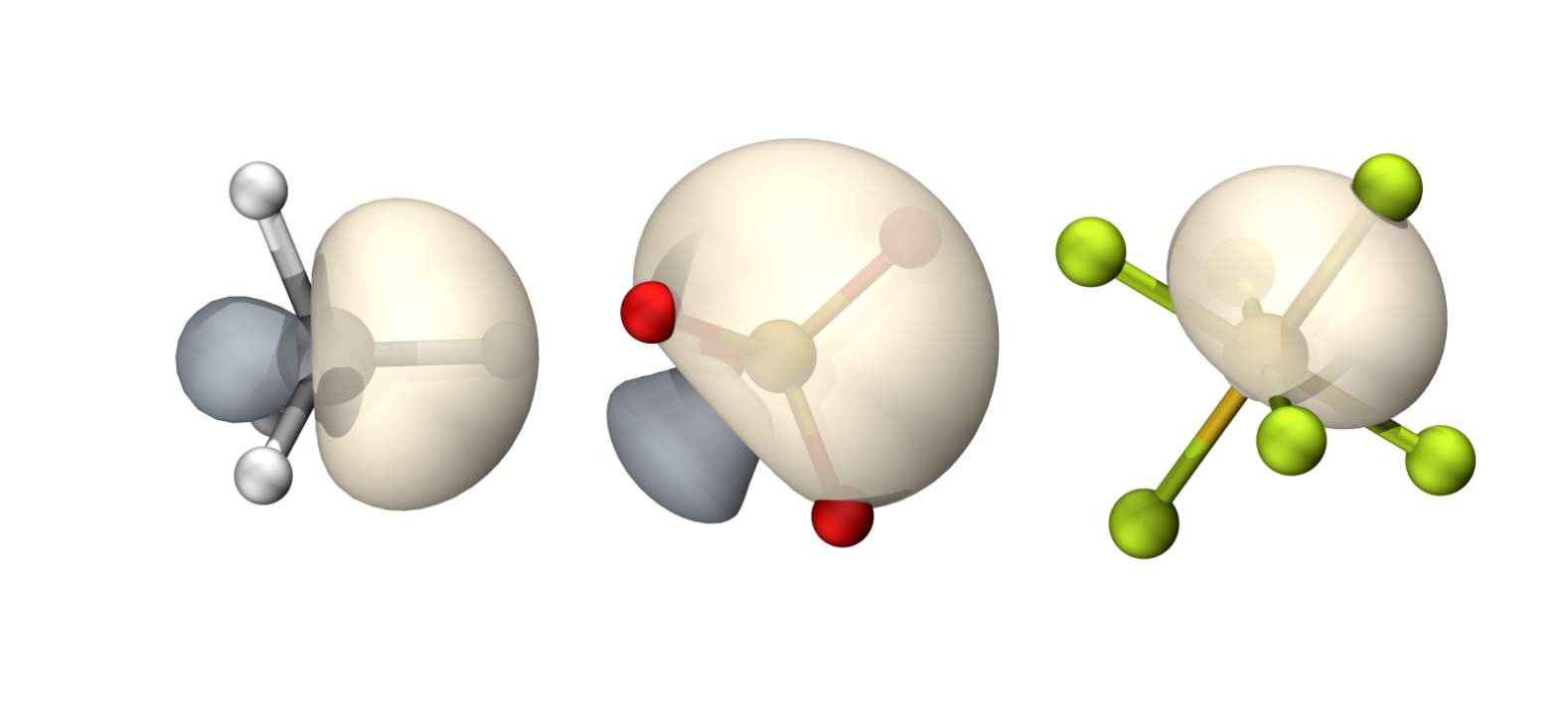
CH4
默认的guess=auto给出的初猜是C的s+px+py+pz轨道,直接str=full轨道不会指向对应的H
这里添加单个共价结构使轨道在优化过程中指向H
CH4
$CTRL
nstr=1 NAO=8 NAE=8 ISCF=5 IPRINT=3
ORBTYP=HAO FRGTYP=SAO
INT=LIBCINT BASIS=def2-TZVP
$END
$FRAG
1*6
SPXPYPZ 1
SPXPYPZ 1
S 2
S 3
S 4
S 5
$END
$ORB
1*9
1
2
3
2
4
2
5
2
6
$END
$str
1 1 2 3 4 5 6 7 8 9
$end
$GEO
C -0.00000000 -0.00000000 -0.00000000
H 0.00000000 0.00000000 1.08943189
H -0.00000000 -1.02712623 -0.36314396
H 0.88951741 0.51356312 -0.36314396
H -0.88951741 0.51356312 -0.36314396
$END碳的轨道具有65.82%的p成分(Mulliken),约sp1.93
s/p之间正交,Mulliken和Stout-Politzer给出的结果一致,SCPA可能低估p成分
full
guess=read读取轨道文件添加str=full进行计算
此时相当于CAS(8,8),共有1764个VB结构。IPRINT=3时.xmo输出高达180MB
结果
轨道:
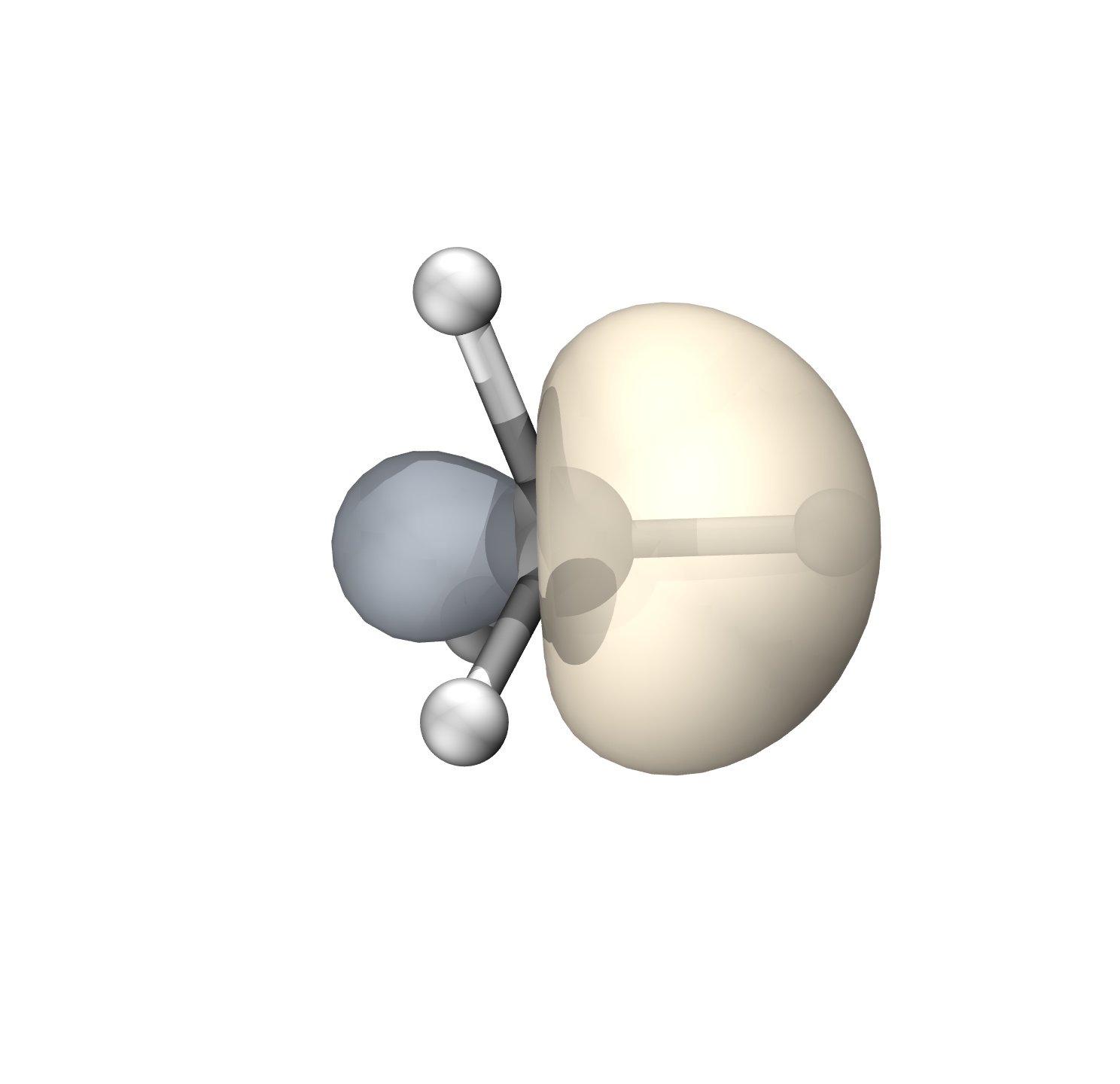
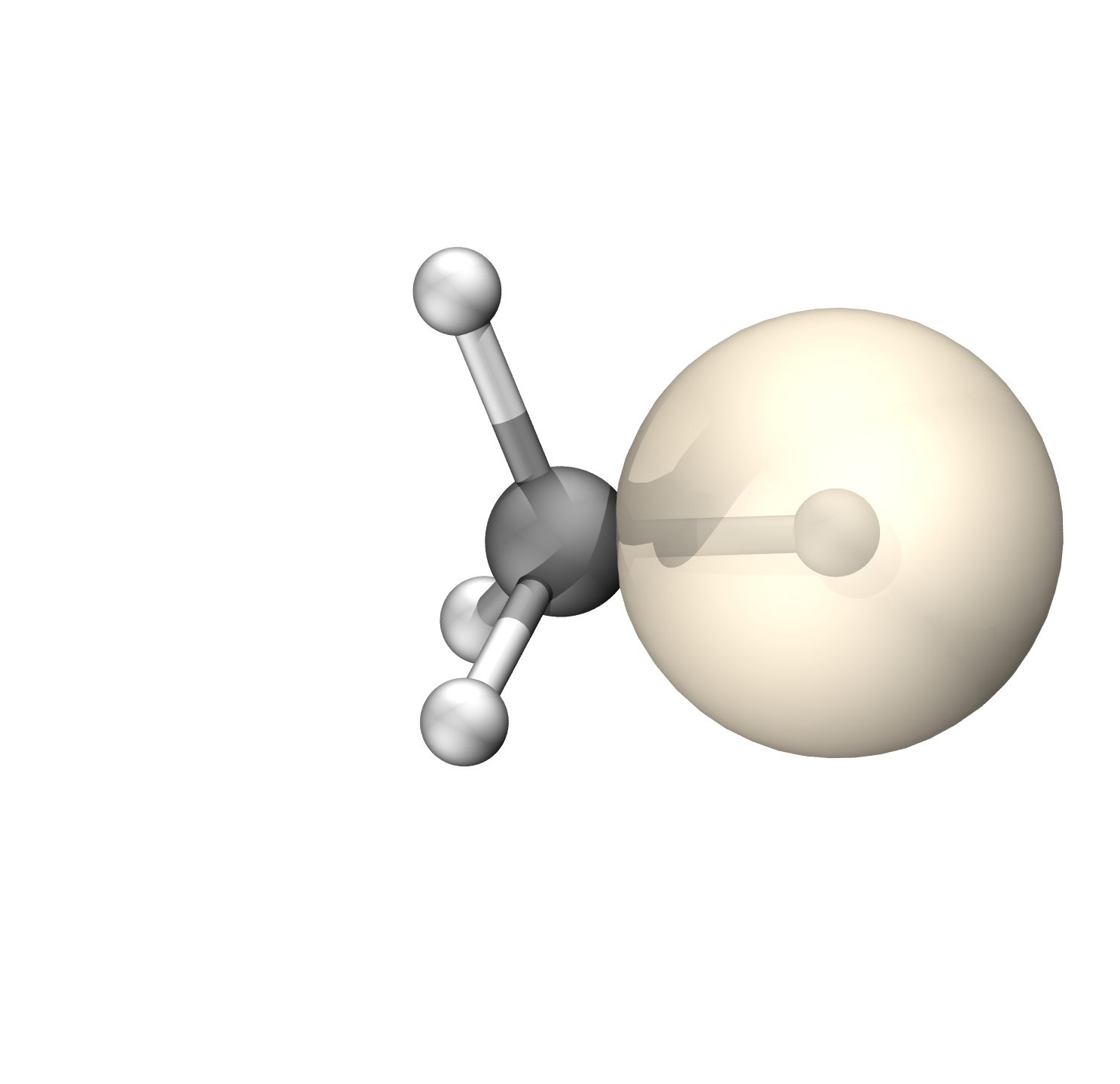
碳的轨道具有70.8%的p成分(Mulliken),约sp2.42
| 结构 | Coulson Chirgwin Weight |
|---|---|
 ×1 ×1 | 18.94% |
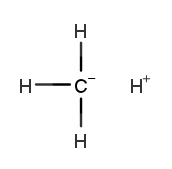 ×4 ×4 | 19.94% |
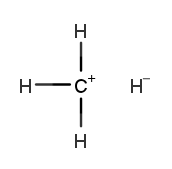 ×4 ×4 | 13.29% |
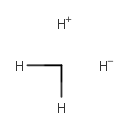 ×12 ×12 | 22.19% |
| 结构 | Renormalized Weight |
|---|---|
| ×1 | 49.68% |
| ×4 | 20.41% |
| ×4 | 9.81% |
| ×12 | 13.21% |
查看输出文件还可以发现这样的结构
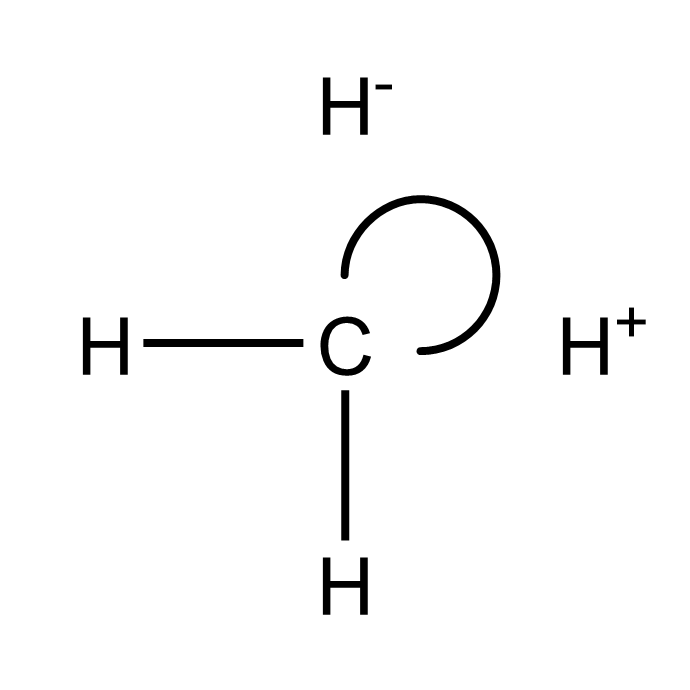
添加极化函数
$frag改为
$FRAG
1*6
SPXPYPZDXYDXZDYZDXXDYYDZZFXXXFYYYFZZZFXXYFXYYFXXZFXZZFYZZFYYZFXYZ 1
SPXPYPZDXYDXZDYZDXXDYYDZZFXXXFYYYFZZZFXXYFXYYFXXZFXZZFYZZFYYZFXYZ 1
SPXPYPZ 2
SPXPYPZ 3
SPXPYPZ 4
SPXPYPZ 5
$END碳的轨道具有67.75%的p成分(Mulliken),约sp1.87
由于6d,10f基函数不正交,可能高估df成分,三种方法给出的结果也不同
CH4光电子能谱
考虑CH4电离一个电子后CH4+的VB结构。最简单的情况可以取四个VB结构
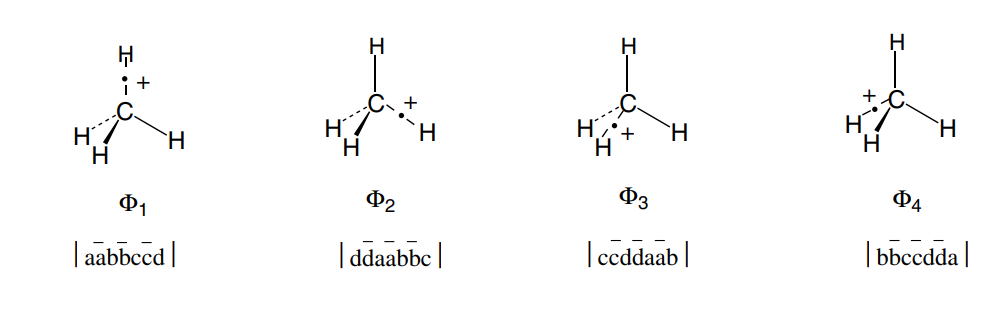
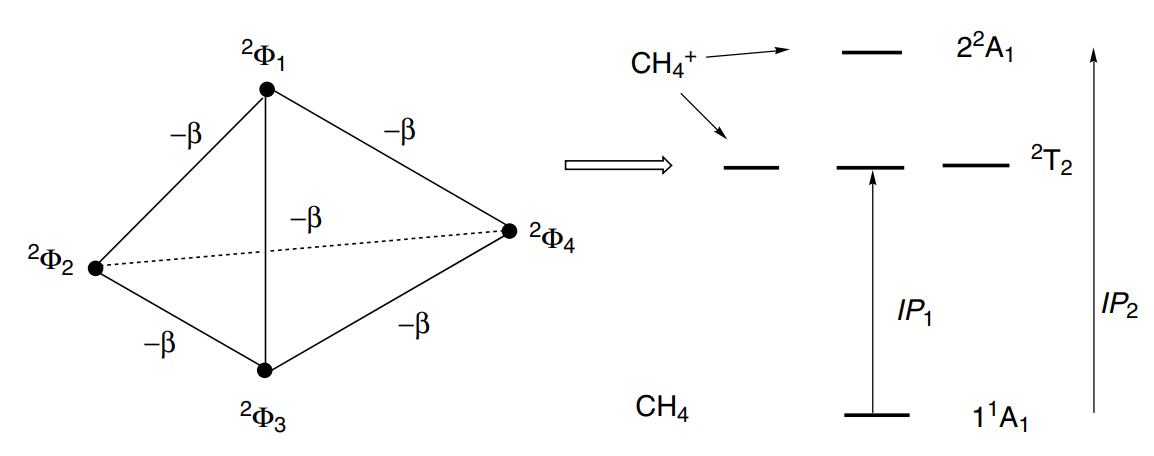
显然这四个结构能量相同,但是VB结构之间具有耦合,无法获取单独的一个结构,哈密顿矩阵为:
对角化之后可以得到一个能量较低高的态和三重简并的能量较低的态,符合光电子能谱中出现的一大一小两个峰
SO4_2-
guess
默认选项guess=auto通过对角化片段Fock矩阵提供初猜
guess=unit将片段内其中一个基函数设为1
对于复杂体系这两种方法无法给出合理初猜
guess=NBO和guess=MO通过NBO和MO构建初猜,不过很难用
NBO轨道可能与VB/MO轨道相去甚远,效果并不好
这里使用PM定域化HF轨道配合XMVB-tools/molden2gus.py提供初猜
轨道使用Gaussian计算,Multiwfn定域化轨道并转换为molden文件
基组为6-311+g(2d,p)(由于基组定义不一致,使用自定义基组,在basis目录下添加自定义基组文件,basis/basis.idx中添加基组名称-文件对应关系)
molden2gus.py输入
1 1,6,7,8,9 # S的内层电子
2 3 # O的内层电子
3 2
4 4
5 5
2 12 # O孤对电子
3 11
4 10
5 13
1,2 18 # 8个 O-S pi轨道
1,3 19
1,4 20
1,5 21
1,2 24
1,3 23
1,4 22
1,5 25
1 15 # O-S sigma 8活性轨道
2 15
1 14
3 14
1 17
4 17
1 16
5 16
q输入
XMVB输入,只取了20个结构
添加了初猜的的输入文件
$CTRL
nstr=20 NAO=8 NAE=8 ISCF=5 IPRINT=3
ORBTYP=HAO FRGTYP=SAO NCHARGE=-2
INT=LIBCINT BASIS=qaq # 自定义6-311+g(2d,p)基组
guess=read
$END
$FRAG
1*5 2*4
SPXPYPZ 1 # S未添加D极化
SPXPYPZDXYDXZDYZDXXDYYDZZ 2
SPXPYPZDXYDXZDYZDXXDYYDZZ 3
SPXPYPZDXYDXZDYZDXXDYYDZZ 4
SPXPYPZDXYDXZDYZDXXDYYDZZ 5
SPXPYPZDXYDXZDYZDXXDYYDZZ 1 2 # pi保持在两个原子上离域
SPXPYPZDXYDXZDYZDXXDYYDZZ 1 3
SPXPYPZDXYDXZDYZDXXDYYDZZ 1 4
SPXPYPZDXYDXZDYZDXXDYYDZZ 1 5
$END
$ORB
1*29
1 # 5个S内层电子
1
1
1
1
2 # 4个O内层电子
3
4
5
2 # O孤对电子
3
4
5
6 # 8个 O-S pi轨道
7
8
9
6
7
8
9
1 # O-S sigma 8活性轨道
2
1
3
1
4
1
5
$END
$str
1:21 22 23 24 25 26 27 28 29 # 共价结构
1:21 22 22 24 25 26 27 28 29 # O+S-一阶离子
1:21 22 23 24 24 26 27 28 29
1:21 22 23 24 25 26 26 28 29
1:21 22 23 24 25 26 27 28 28
1:21 23 23 24 25 26 27 28 29 # O-S+一阶离子
1:21 22 23 25 25 26 27 28 29
1:21 22 23 24 25 27 27 28 29
1:21 22 23 24 25 26 27 29 29
1:21 23 23 25 25 26 27 28 29 # O-S+二阶离子
1:21 23 23 24 25 27 27 28 29
1:21 23 23 24 25 26 27 29 29
1:21 22 23 25 25 27 27 28 29
1:21 22 23 25 25 26 27 29 29
1:21 22 23 24 25 27 27 29 29
1:21 23 23 25 25 27 27 28 29 # O-S+三阶离子
1:21 23 23 25 25 26 27 29 29
1:21 23 23 24 25 27 27 29 29
1:21 22 23 25 25 27 27 29 29
1:21 23 23 25 25 27 27 29 29 # O-S+四阶离子
$end
$GEO
S 0.000000 0.000000 0.000000
O 0.867917 0.867917 0.867917
O -0.867917 -0.867917 0.867917
O -0.867917 0.867917 -0.867917
O 0.867917 -0.867917 -0.867917
$END
大基组下VB计算很难收敛,建议将itmax调大,如未收敛则读取最后的轨道文件继续计算
结果
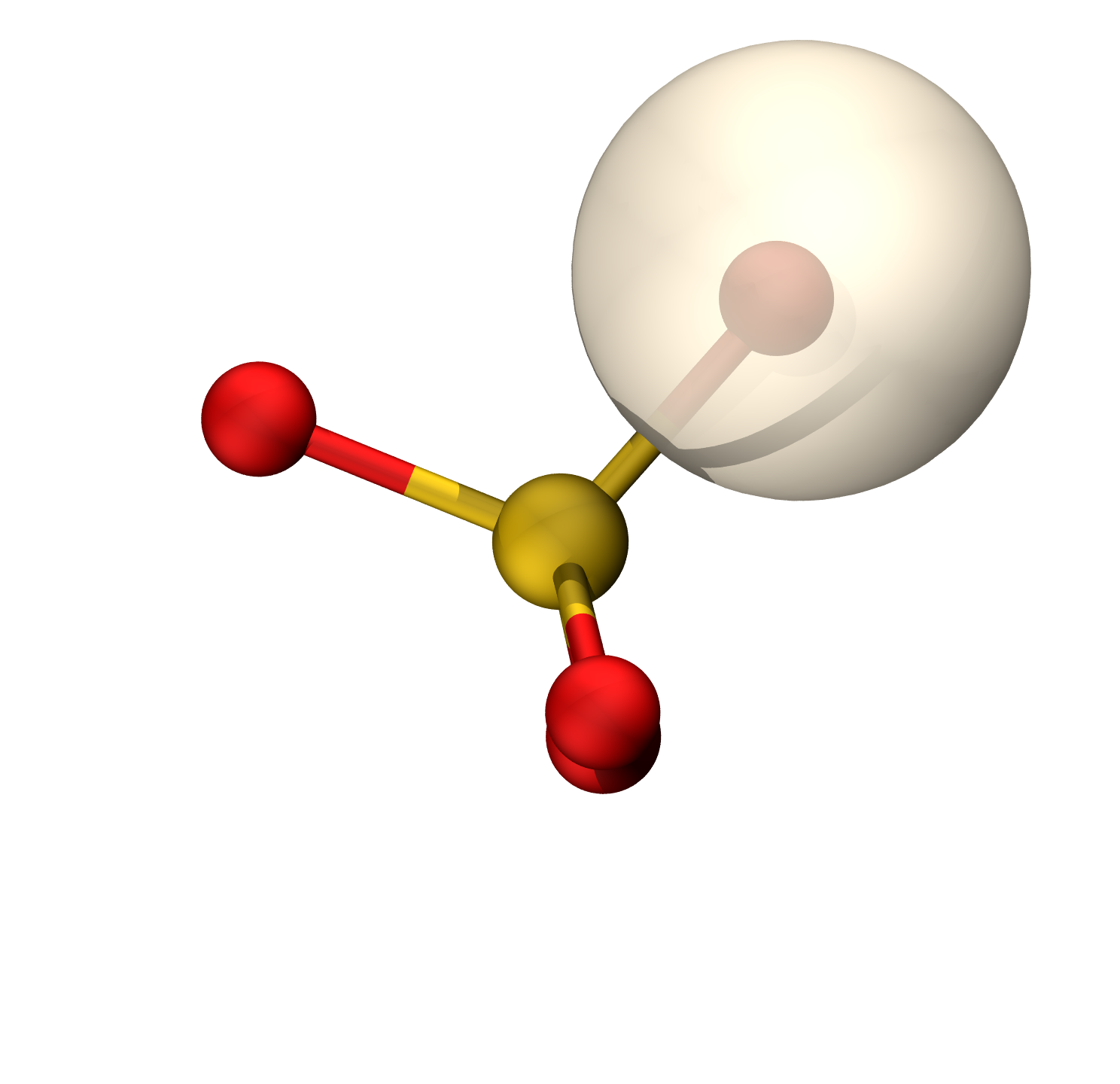
硫的轨道具有38.82%的p成分(Mulliken),约sp0.63
等值面0.06
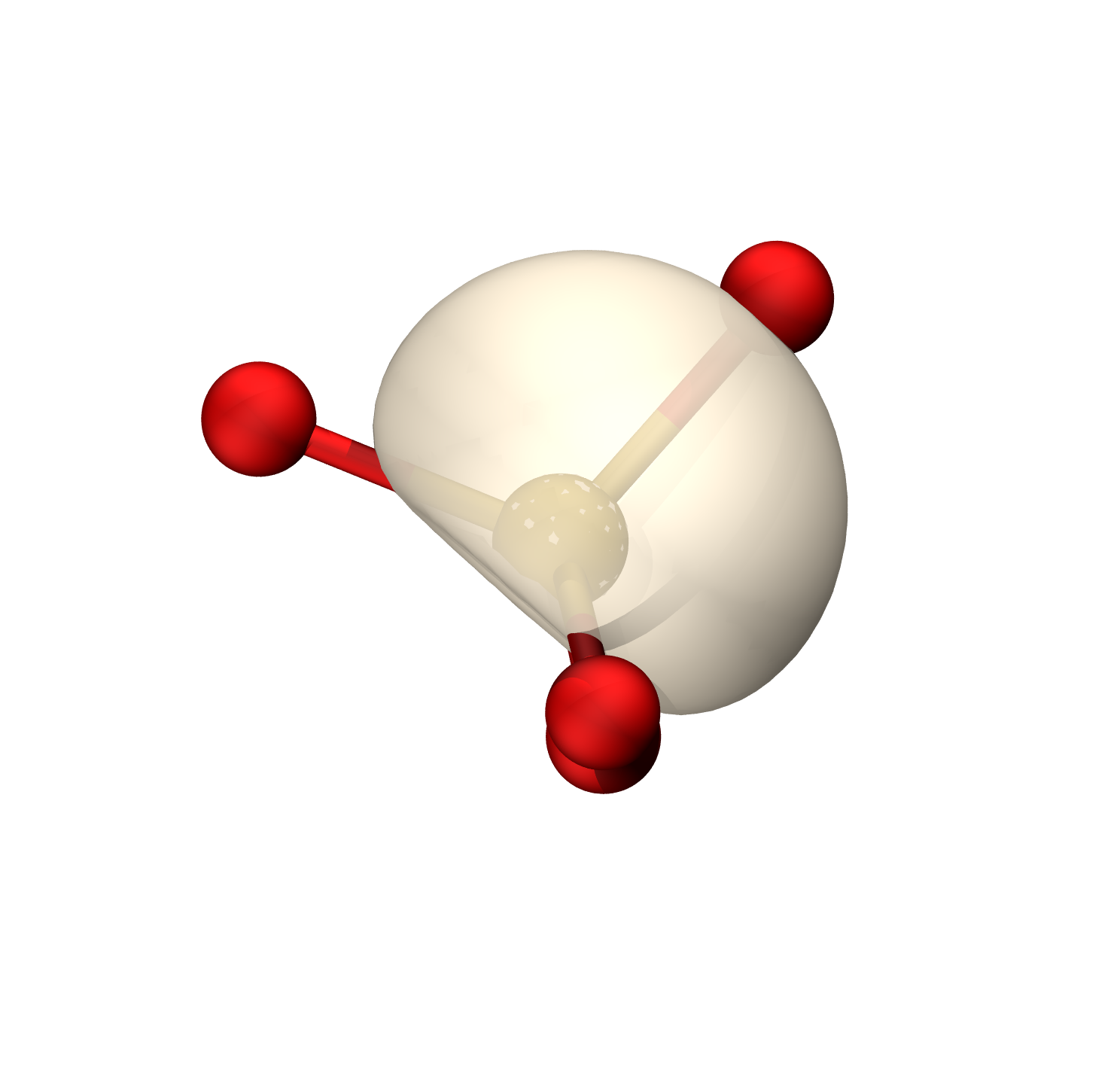
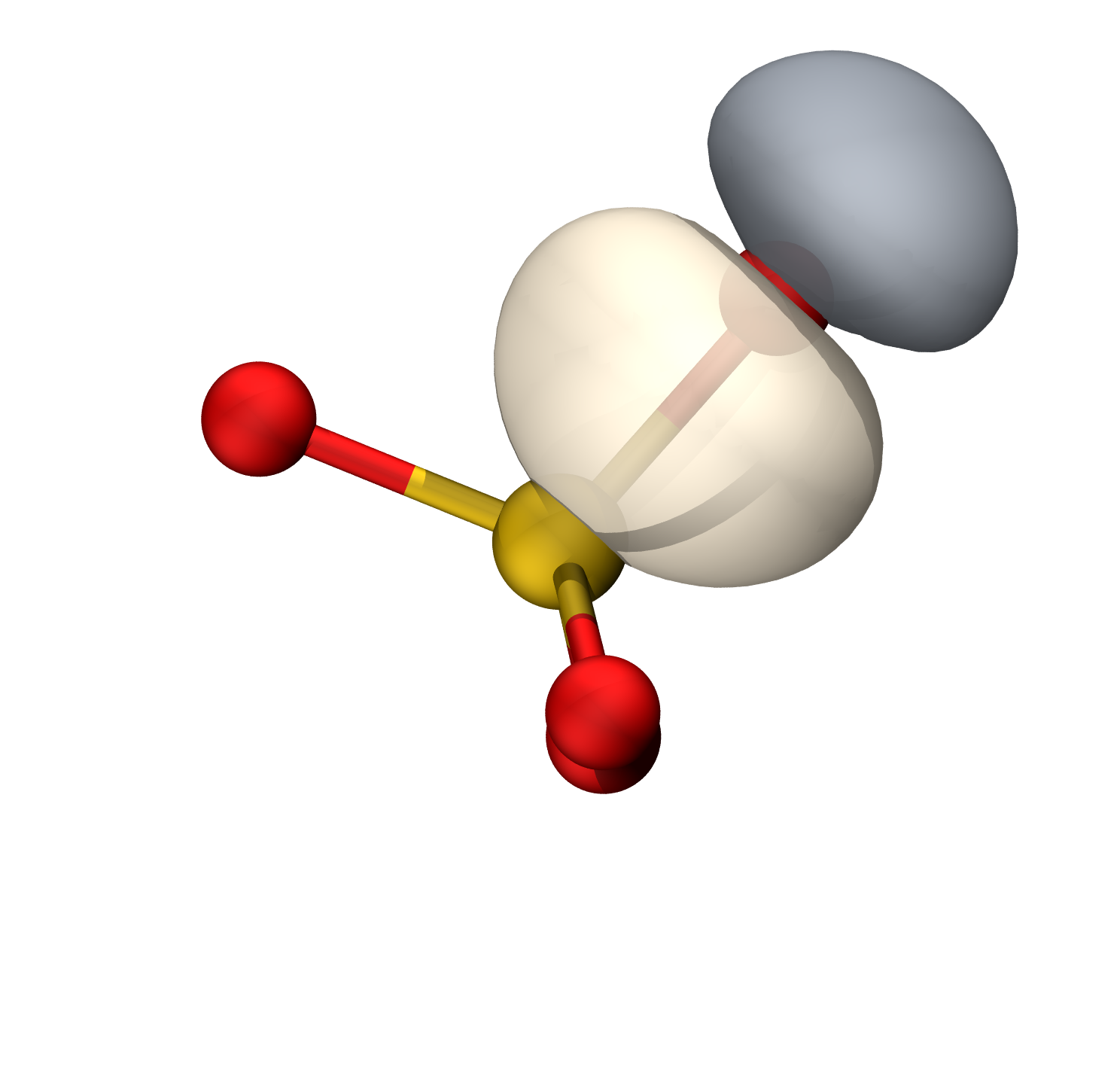
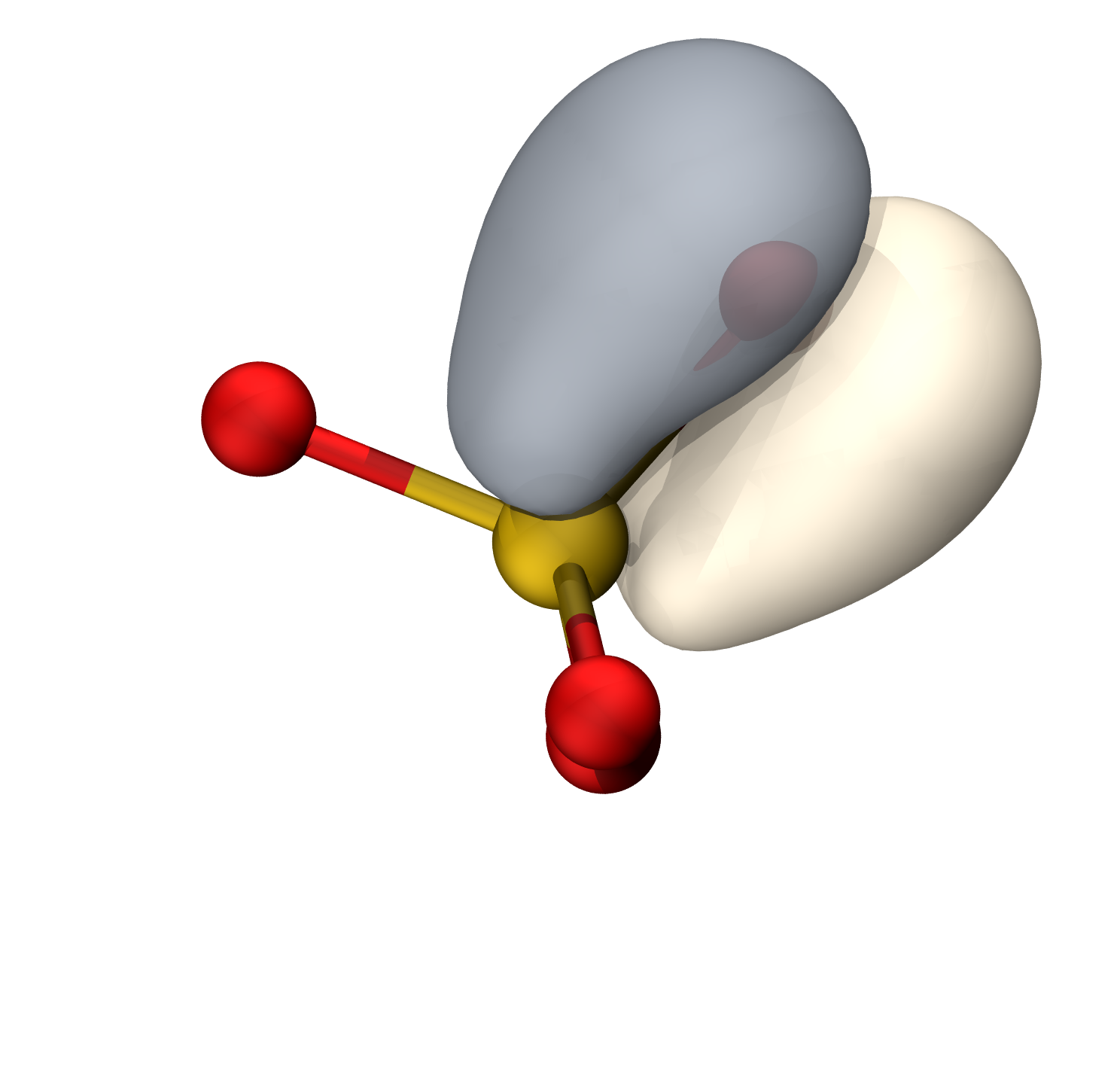
O孤对
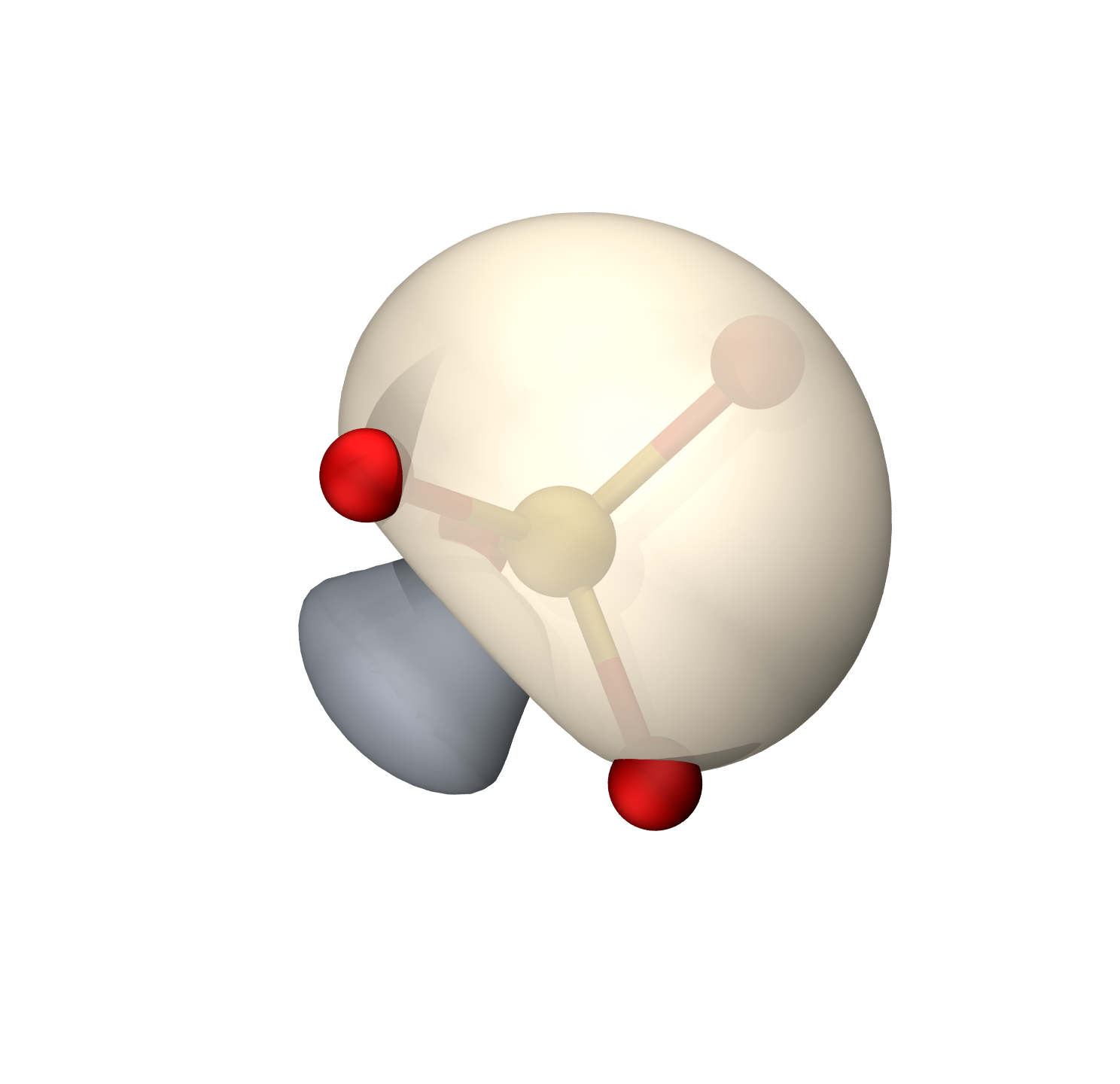
S-O π
等值面0.009
相关内容可以参考zhihu回答
| 结构 | Coulson Chirgwin Weight |
|---|---|
| 共价×1 | 3.60% |
| O+S-一阶离子×4 | 3.49% |
| O-S+一阶离子×4 | 33.79% |
| O-S+二阶离子×6 | 41.02% |
| O-S+三阶离子×4 | 17.00% |
| O-S+四阶离子×1 | 1.07% |
| 结构 | Renormalized Weight |
|---|---|
| 共价×1 | 1.68% |
| O+S-一阶离子×4 | 1.15% |
| O-S+一阶离子×4 | 40.57% |
| O-S+二阶离子×6 | 43.93% |
| O-S+三阶离子×4 | 12.44% |
| O-S+四阶离子×1 | 0.21% |
添加D极化之后p成分(Mulliken)42%,约sp0.8
SF6
活性空间为12,12,总结构数量22.6万,这里使用脚本生成了70个结构
n阶F-S+离子结构相当于
SF6
$CTRL
nstr=70 NAO=12 NAE=12 ISCF=5 IPRINT=3
ORBTYP=HAO FRGTYP=SAO
INT=LIBCINT BASIS=6-311g*
guess=read
$END
$FRAG
1*7 2*12 1*6
SPXPYPZDXYDXZDYZDXXDYYDZZ 1
SPXPYPZDXYDXZDYZDXXDYYDZZ 2
SPXPYPZDXYDXZDYZDXXDYYDZZ 3
SPXPYPZDXYDXZDYZDXXDYYDZZ 4
SPXPYPZDXYDXZDYZDXXDYYDZZ 5
SPXPYPZDXYDXZDYZDXXDYYDZZ 6
SPXPYPZDXYDXZDYZDXXDYYDZZ 7
PXDXZDYZ 2 1 # pi保持在两个原子上离域
PXDXZDYZ 6 1
PYDXZDYZ 2 1
PYDXZDYZ 6 1
PXDXYDYZ 3 1
PXDXYDYZ 5 1
PZDXYDYZ 3 1
PZDXYDYZ 5 1
PYDXYDXZ 4 1
PYDXYDXZ 7 1
PZDXYDXZ 4 1
PZDXYDXZ 7 1
SPZDXXDYYDZZ 2 # F的s孤对
SPZDXXDYYDZZ 6
SPYDXXDYYDZZ 3
SPYDXXDYYDZZ 5
SPXDXXDYYDZZ 4
SPXDXXDYYDZZ 7
$END
$ORB
1*41
1 # 5个S内层电子
1
1
1
1
2 # 6个F内层电子
3
4
5
6
7
8 # 12个F-S pi
9
10
11
12
13
14
15
16
17
18
19
20 # 6个F孤对电子
21
22
23
24
25
1 # F-S sigma 8活性轨道
2
1
3
1
4
1
5
1
6
1
7
$END
$str
1:29 30 31 32 33 34 35 36 37 38 39 40 41 # 共价结构
1:29 31 31 32 33 34 35 36 37 38 39 40 41 # F-S+一阶离子
1:29 30 31 33 33 34 35 36 37 38 39 40 41
1:29 30 31 32 33 35 35 36 37 38 39 40 41
1:29 30 31 32 33 34 35 37 37 38 39 40 41
1:29 30 31 32 33 34 35 36 37 39 39 40 41
1:29 30 31 32 33 34 35 36 37 38 39 41 41
1:29 30 30 32 33 34 35 36 37 38 39 40 41 # F+S-一阶离子
1:29 30 31 32 32 34 35 36 37 38 39 40 41
1:29 30 31 32 33 34 34 36 37 38 39 40 41
1:29 30 31 32 33 34 35 36 36 38 39 40 41
1:29 30 31 32 33 34 35 36 37 38 38 40 41
1:29 30 31 32 33 34 35 36 37 38 39 40 40
1:29 31 31 33 33 34 35 36 37 38 39 40 41 # F-S+二阶离子
1:29 31 31 32 33 35 35 36 37 38 39 40 41
1:29 31 31 32 33 34 35 37 37 38 39 40 41
1:29 31 31 32 33 34 35 36 37 39 39 40 41
1:29 31 31 32 33 34 35 36 37 38 39 41 41
1:29 30 31 33 33 35 35 36 37 38 39 40 41
1:29 30 31 33 33 34 35 37 37 38 39 40 41
1:29 30 31 33 33 34 35 36 37 39 39 40 41
1:29 30 31 33 33 34 35 36 37 38 39 41 41
1:29 30 31 32 33 35 35 37 37 38 39 40 41
1:29 30 31 32 33 35 35 36 37 39 39 40 41
1:29 30 31 32 33 35 35 36 37 38 39 41 41
1:29 30 31 32 33 34 35 37 37 39 39 40 41
1:29 30 31 32 33 34 35 37 37 38 39 41 41
1:29 30 31 32 33 34 35 36 37 39 39 41 41
1:29 31 31 33 33 35 35 36 37 38 39 40 41 # F-S+三阶离子
1:29 31 31 33 33 34 35 37 37 38 39 40 41
1:29 31 31 33 33 34 35 36 37 39 39 40 41
1:29 31 31 33 33 34 35 36 37 38 39 41 41
1:29 31 31 32 33 35 35 37 37 38 39 40 41
1:29 31 31 32 33 35 35 36 37 39 39 40 41
1:29 31 31 32 33 35 35 36 37 38 39 41 41
1:29 31 31 32 33 34 35 37 37 39 39 40 41
1:29 31 31 32 33 34 35 37 37 38 39 41 41
1:29 31 31 32 33 34 35 36 37 39 39 41 41
1:29 30 31 33 33 35 35 37 37 38 39 40 41
1:29 30 31 33 33 35 35 36 37 39 39 40 41
1:29 30 31 33 33 35 35 36 37 38 39 41 41
1:29 30 31 33 33 34 35 37 37 39 39 40 41
1:29 30 31 33 33 34 35 37 37 38 39 41 41
1:29 30 31 33 33 34 35 36 37 39 39 41 41
1:29 30 31 32 33 35 35 37 37 39 39 40 41
1:29 30 31 32 33 35 35 37 37 38 39 41 41
1:29 30 31 32 33 35 35 36 37 39 39 41 41
1:29 30 31 32 33 34 35 37 37 39 39 41 41
1:29 31 31 33 33 35 35 37 37 38 39 40 41 # F-S+四阶离子
1:29 31 31 33 33 35 35 36 37 39 39 40 41
1:29 31 31 33 33 35 35 36 37 38 39 41 41
1:29 31 31 33 33 34 35 37 37 39 39 40 41
1:29 31 31 33 33 34 35 37 37 38 39 41 41
1:29 31 31 33 33 34 35 36 37 39 39 41 41
1:29 31 31 32 33 35 35 37 37 39 39 40 41
1:29 31 31 32 33 35 35 37 37 38 39 41 41
1:29 31 31 32 33 35 35 36 37 39 39 41 41
1:29 31 31 32 33 34 35 37 37 39 39 41 41
1:29 30 31 33 33 35 35 37 37 39 39 40 41
1:29 30 31 33 33 35 35 37 37 38 39 41 41
1:29 30 31 33 33 35 35 36 37 39 39 41 41
1:29 30 31 33 33 34 35 37 37 39 39 41 41
1:29 30 31 32 33 35 35 37 37 39 39 41 41
1:29 31 31 33 33 35 35 37 37 39 39 40 41 # F-S+五阶离子
1:29 31 31 33 33 35 35 37 37 38 39 41 41
1:29 31 31 33 33 35 35 36 37 39 39 41 41
1:29 31 31 33 33 34 35 37 37 39 39 41 41
1:29 31 31 32 33 35 35 37 37 39 39 41 41
1:29 30 31 33 33 35 35 37 37 39 39 41 41
1:29 31 31 33 33 35 35 37 37 39 39 41 41 # F-S+六阶离子
$end
$gus
xxx
$end
$GEO
S 0.000000 0.000000 0.000000
F 0.000000 0.000000 1.565367
F 0.000000 1.565367 0.000000
F -1.565367 0.000000 0.000000
F 0.000000 -1.565367 0.000000
F 0.000000 0.000000 -1.565367
F 1.565367 0.000000 0.000000
$END结果
硫的轨道具有42.93%的p成分(Mulliken),约sp0.95
等值面0.06
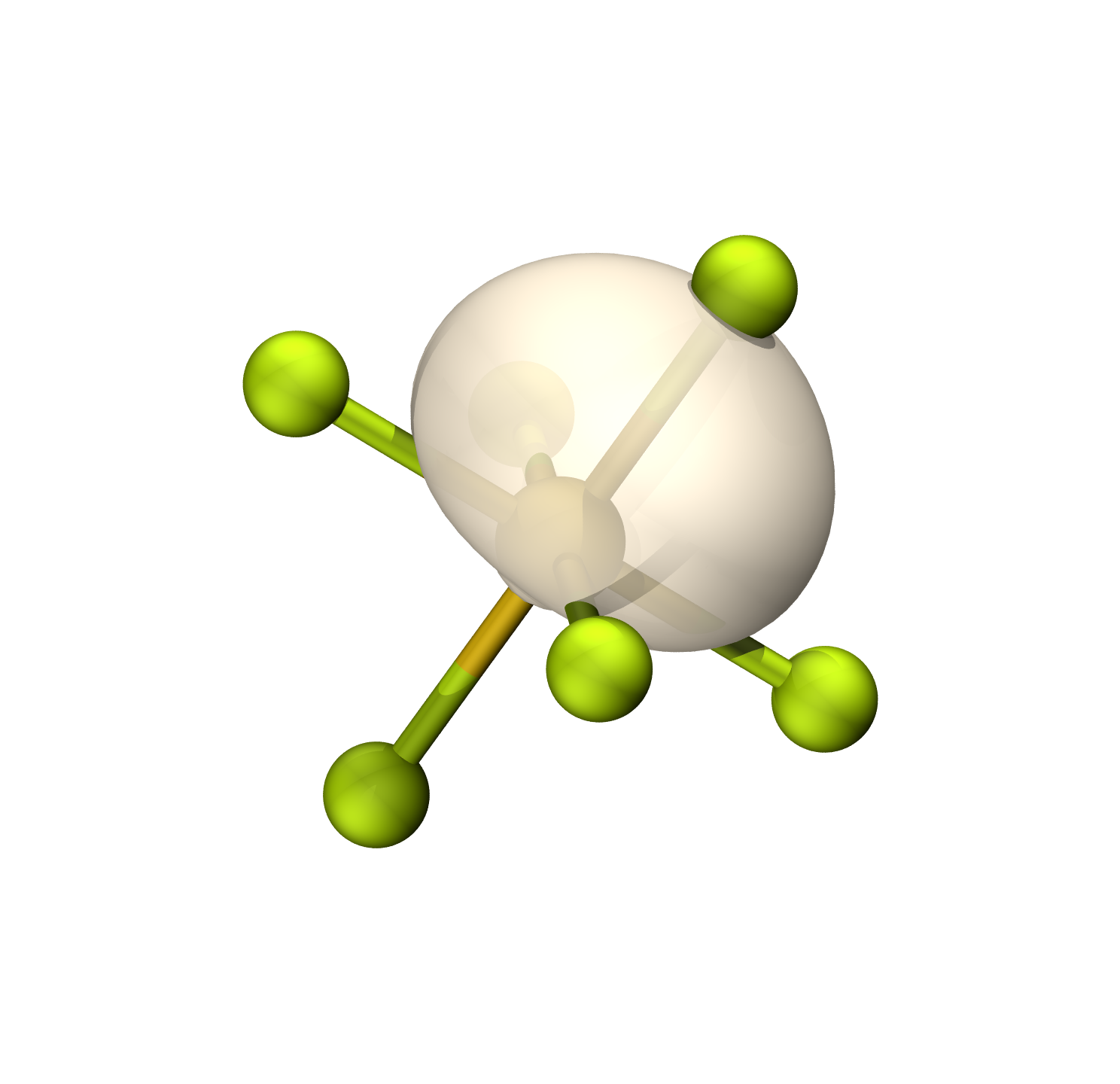
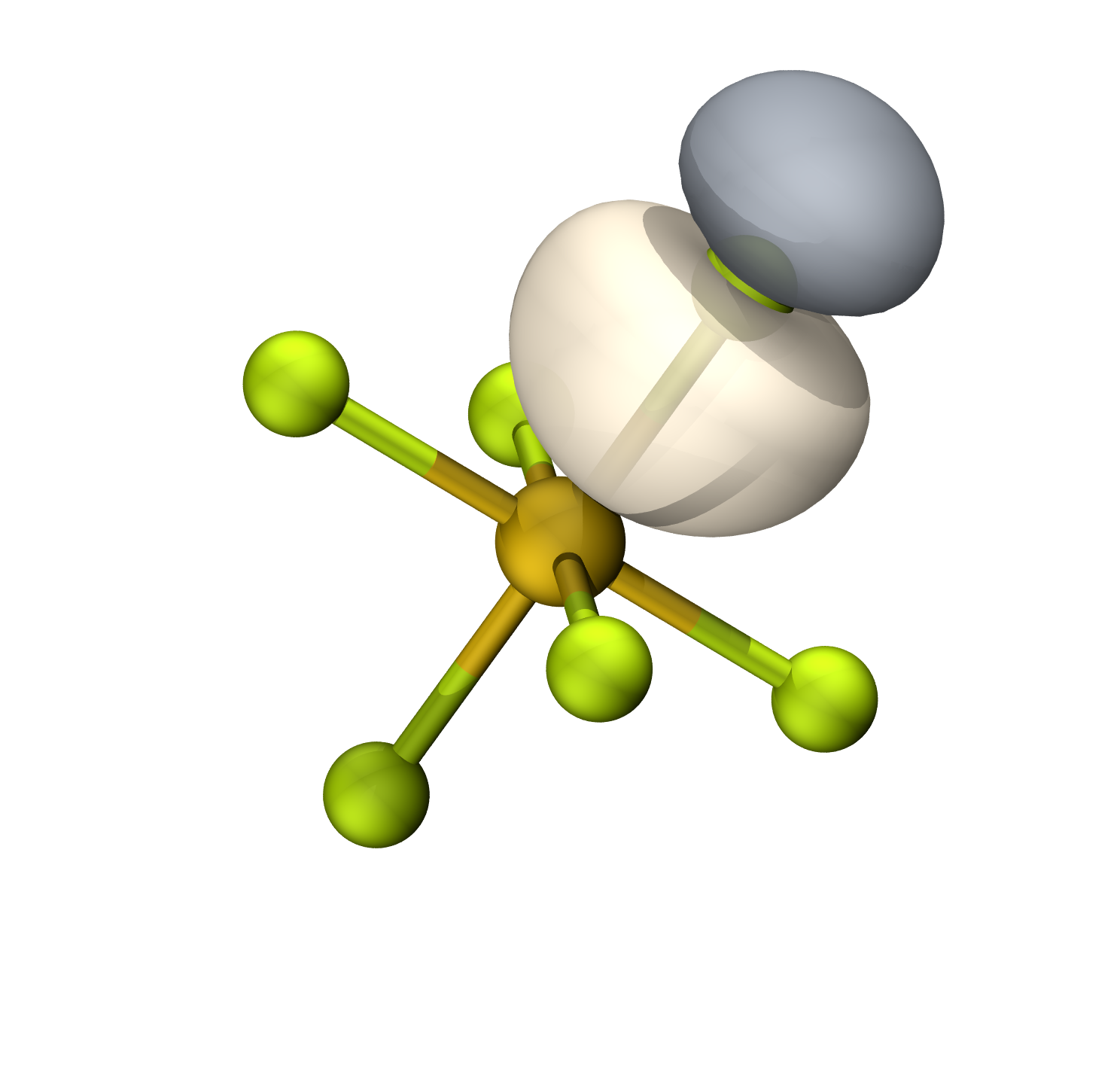
F孤对
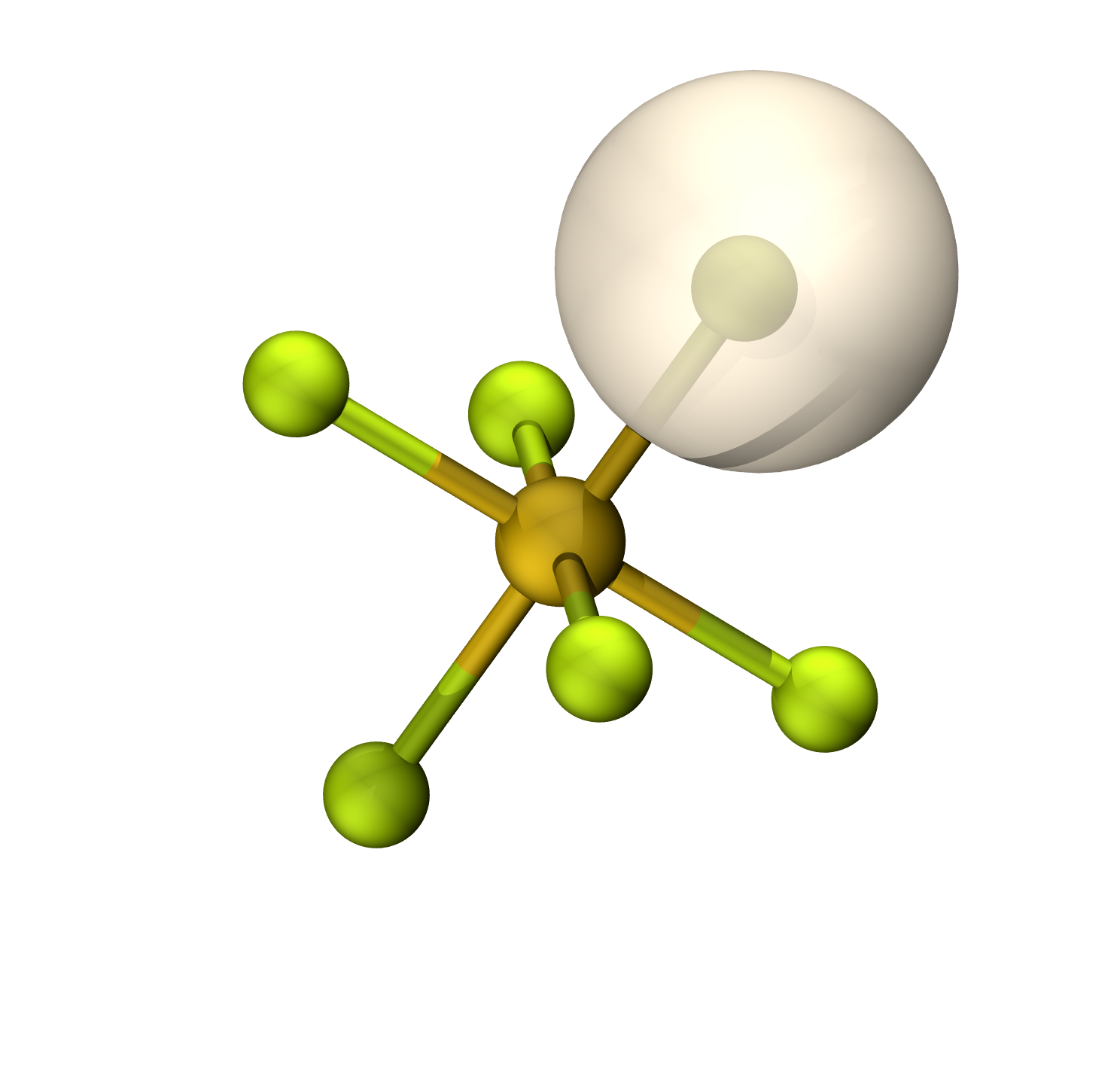
S-F π

| 结构 | Coulson Chirgwin Weight |
|---|---|
| 共价×1 | -0.44% |
| F+S-一阶离子×6 | 1.38% |
| F-S+一阶离子×6 | 11.84% |
| F-S+二阶离子×15 | 32.51% |
| F-S+三阶离子×20 | 36.25% |
| F-S+四阶离子×15 | 16.12% |
| F-S+五阶离子×6 | 2.3% |
| F-S+六阶离子×1 | 0.01% |
| 结构 | Renormalized Weight |
|---|---|
| 共价×1 | 0.08% |
| F+S-一阶离子×6 | 0.56% |
| F-S+一阶离子×6 | 11.28% |
| F-S+二阶离子×15 | 37.21% |
| F-S+三阶离子×20 | 38.64% |
| F-S+四阶离子×15 | 11.53% |
| F-S+五阶离子×6 | 0.67% |
| F-S+六阶离子×1 | 0.0001% |
使用软件:
可视化轨道: VMD vcube
VB计算: XMVB
转换XMVB轨道文件 XMVB-tools
分析轨道、计算轨道cube文件: Multiwfn
建议阅读:
Orbital Hybridization in Modern Valence Bond Wave Functions: Methane, Ethylene, and Acetylene