
分子轨道理论的处理
从分子轨道理论出发的环丁二烯HOMO轨道二重简并,几何结构变形为以降低其中一个态的能量

这导致一个小的HOMO-LUMO gap。在CTOCD中,较小的gap以及两个轨道的对称性导致强顺磁环电流,反芳香性(环丁二烯的不稳定性可能还有别的因素)
价键理论的失败(?)
VB中环丁二烯可以画出与苯类似的共振式,所以环丁二烯也具有芳香性(?)
计算
D4h 仅共价结构
使用str=cov生成2种共价结构
$ctrl
str=cov nao=4 nae=4 iscf=5 iprint=3
orbtyp=hao frgtyp=sao
int=libcint basis=cc-pvdz
$end
$frag
8 2*4
spxpydxxdyydzzdxy 1-8
pzdxzdyz 1 5
pzdxzdyz 2 6
pzdxzdyz 3 7
pzdxzdyz 4 8
$end
$orb
1*12 1*4
1
1
1
1
1
1
1
1
1
11
1
1
2
3
4
5
$end
$geo
C -0.71894821 0.71894821 -0.00000000
C -0.71894821 -0.71894821 -0.00000000
C 0.71894821 -0.71894821 -0.00000000
C 0.71894821 0.71894821 -0.00000000
H -1.48149821 1.48149821 0.00000000
H -1.48149821 -1.48149821 0.00000000
H 1.48149821 -1.48149821 0.00000000
H 1.48149821 1.48149821 0.00000000
$end得到能量: -153.66281039
D2h 仅共价结构
将D4h输入文件的$geo替换为D2h结构
$geo
C -0.78845714 0.66510000 0.00000000
C -0.78845714 -0.66510000 0.00000000
C 0.78845714 -0.66510000 0.00000000
C 0.78845714 0.66510000 -0.00000000
H -1.55045714 1.42980000 0.00000000
H -1.55045714 -1.42980000 0.00000000
H 1.55045714 -1.42980000 0.00000000
H 1.55045714 1.42980000 -0.00000000
$end得到能量: -153.65571533
结构反而比低了4.45kcal/mol
可以看出仅有共价结构的VB确实不能得到正确结果
D4h full
将str=cov改为str=full生成所有20种VB结构
查看输出的结构权重可以发现有4个1阶对角离子项,2个2阶离子项都为0(?)
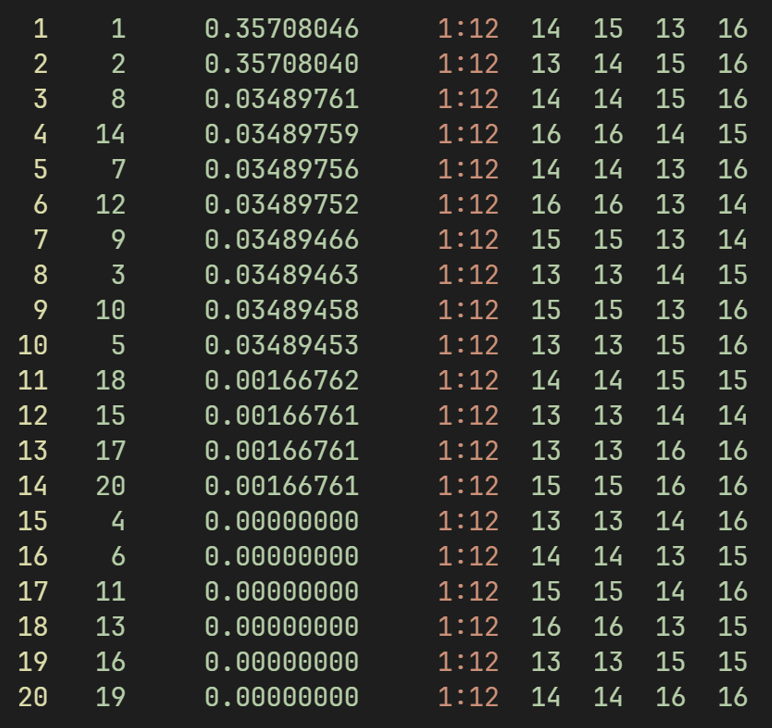 能量: -153.69393178
能量: -153.69393178
D2h full
将D4h full输入文件的$geo替换为D2h结构
此时对角离子结构权重不为0:
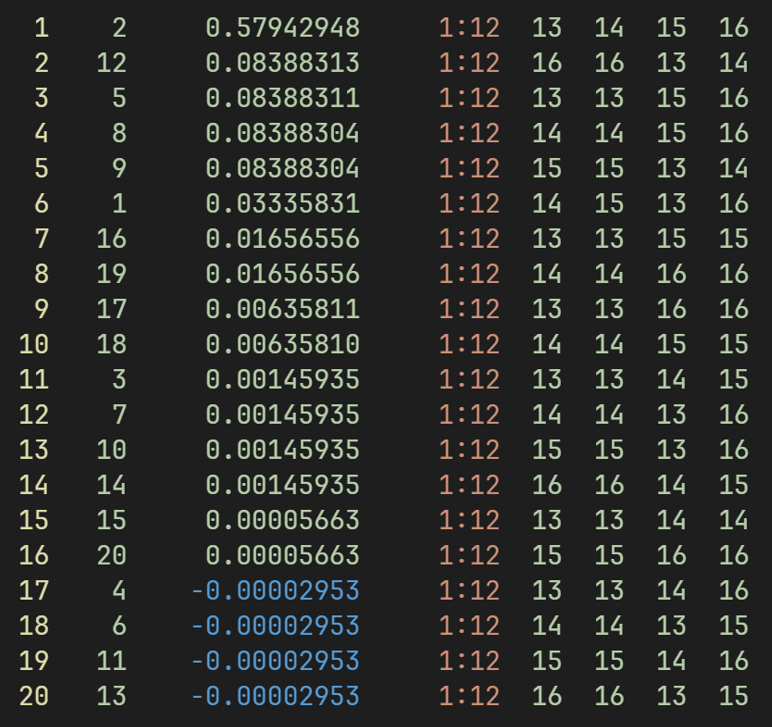 能量: -153.70887144
能量: -153.70887144
结构比低9.37 kcal/mol
加入离子结构的VB给出了正确结果
可见离子结构的重要性
环丁二烯与苯的区别
在苯中为负,基态是,环丁二烯中则相反
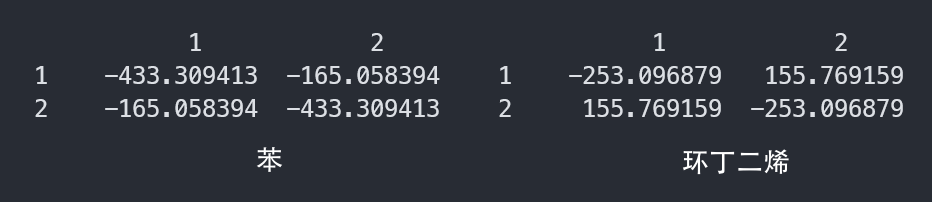

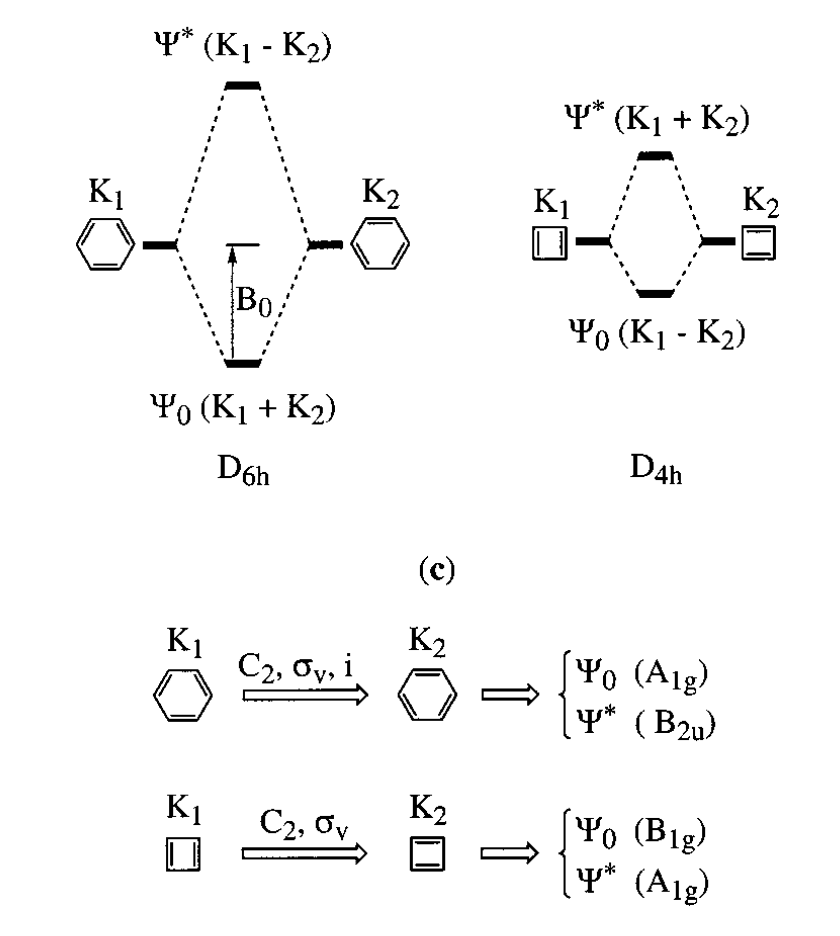
苯中所有一阶离子项都具有基态组合,而环丁二烯的4个1阶对角离子项则没有组合,不能混入基态
(需要注意和轴的方向)
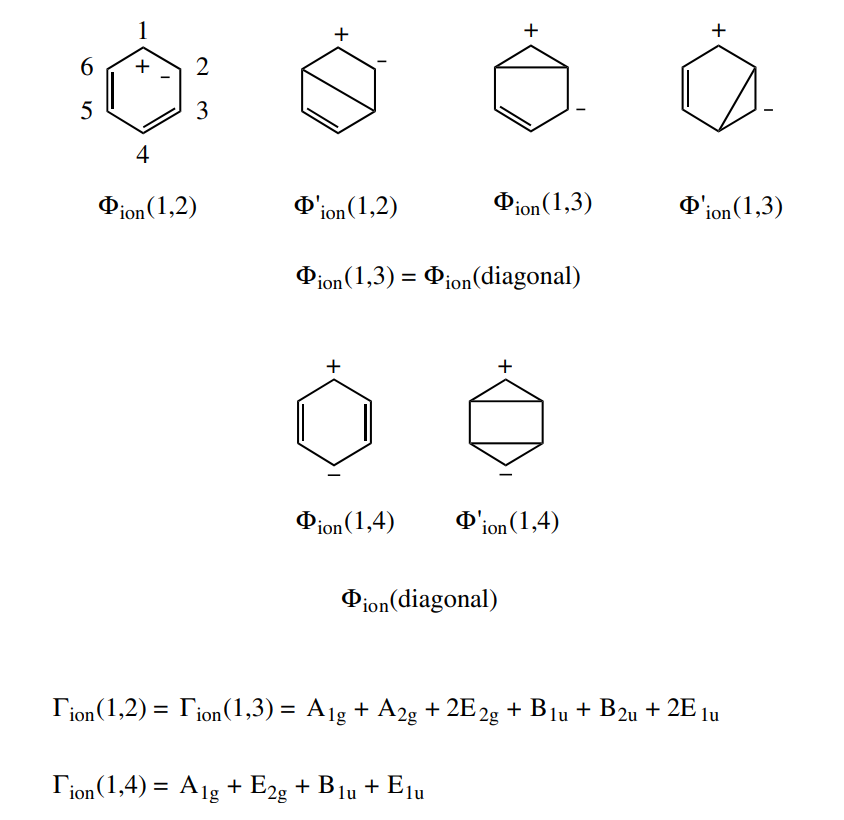
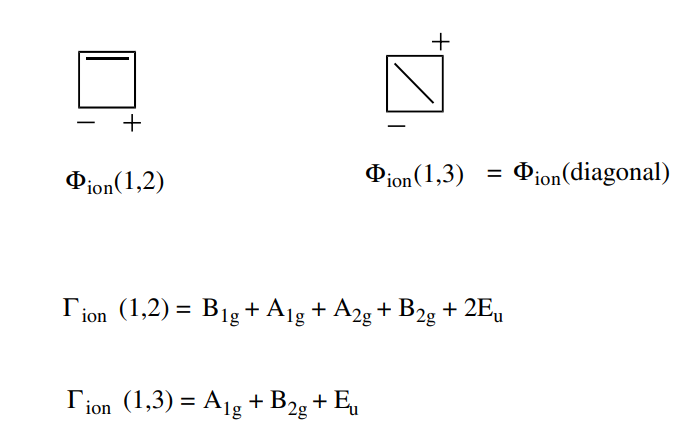
和能量差:
| 结构 | 能量差 kcal/mol |
|---|---|
| cov | -4.45 |
| full | 9.37 |
| 14结构(去除无法混入的一阶离子项) | 7.61 |
| ion(1-2) | 21.5 |
| 仅8个非对角一阶离子项 | 34.18 |
| CASSCF(2,2) | 11.53 |
| CASSCF(4,4) | 5.46 |
所有计算结果的电子能量都是
~~但核排斥~
参考文献