
VB理论
可以参考 XMVB手册 A Chemist’s Guide to Valence Bond Theory
VB理论中术语与MO有对应关系
VBSCFMCSCF
VB结构自旋匹配组态
VB行列式行列式
MO中CI能量通过对角化CI矩阵得到。VBSCF能量也需要对角化VB行列式/VB结构为基的CI矩阵,由于VB结构之间不正交,需要求解:
这和MO中非正交AO基Hartree-Fock的情况一致
为了消掉重叠矩阵,一般使用对称正交化,正交化过程这里不过多赘述,可以参考Szabo Modern Quantum Chemistry:
总之我们需要找出一个矩阵使得,在对称正交化中
然后将变换:
最后求解
对角化后得到能量矩阵,原系数矩阵
为正交矩阵,满足
满足
实际计算中不需要求出所有态的系数和能量
MO理论中计算分子轨道成分时由于基函数不正交,有多种计算AO占比的方法
在VB理论中VB结构不正交,结构权重也有多种划分方法,最常见的是
这和MO中的划分方法一致
VB结构
理想情况下如果有2n个活性轨道,2n个活性电子,可以画出种VB结构
但是能画出的VB结构的数量比自旋匹配组态的数量(CAS(m,n)的CSF数量为)还要多,导致线性相关
为了找全所有线性无关的VB结构,需要使用Rumer规则:
将所有轨道(任意顺序)排列在一圆上,只取连线不相交的结构
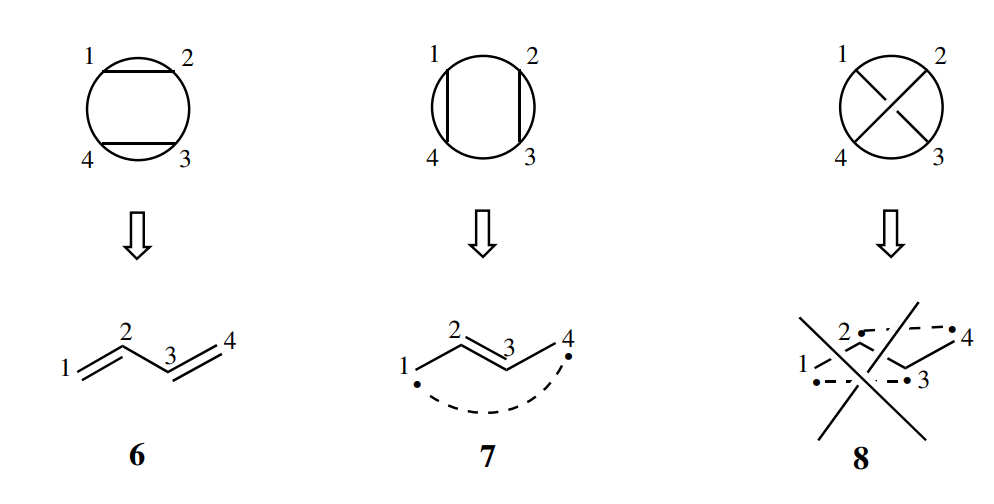
苯
这里结构和基组与官网上的例子完全相同,方便对比
C6H6
$ctrl
str=FULL nao=6 nae=6 iscf=5 iprint=3
orbtyp=hao frgtyp=sao
int=libcint basis=cc-pvdz
$end
$frag
12 2*6
spxpydxxdyydzzdxy 1-12
pzdxzdyz 1 2
pzdxzdyz 3 4
pzdxzdyz 5 6
pzdxzdyz 7 8
pzdxzdyz 9 10
pzdxzdyz 11 12
$end
$orb
1*18 1*6
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
2
3
4
5
6
7
$end
$geo
C 0.6995000584 1.2115696411 0.0000000000
H 1.2460106991 2.1581538376 0.0000000000
C -0.6995000584 1.2115696411 0.0000000000
H -1.2460106991 2.1581538376 0.0000000000
C -1.3990001169 0.0000000000 0.0000000000
H -2.4920213982 0.0000000000 0.0000000000
C -0.6995000584 -1.2115696411 0.0000000000
H -1.2460106991 -2.1581538376 0.0000000000
C 0.6995000584 -1.2115696411 0.0000000000
H 1.2460106991 -2.1581538376 0.0000000000
C 1.3990001169 0.0000000000 0.0000000000
H 2.4920213982 0.0000000000 0.0000000000
$endstr=full表示自动生成所有VB结构,此时不需要nstr=关键词和$str部分
计算苯时我们只使用6个pz轨道和6个π电子,nao=6 nae=6
VB结构共175个
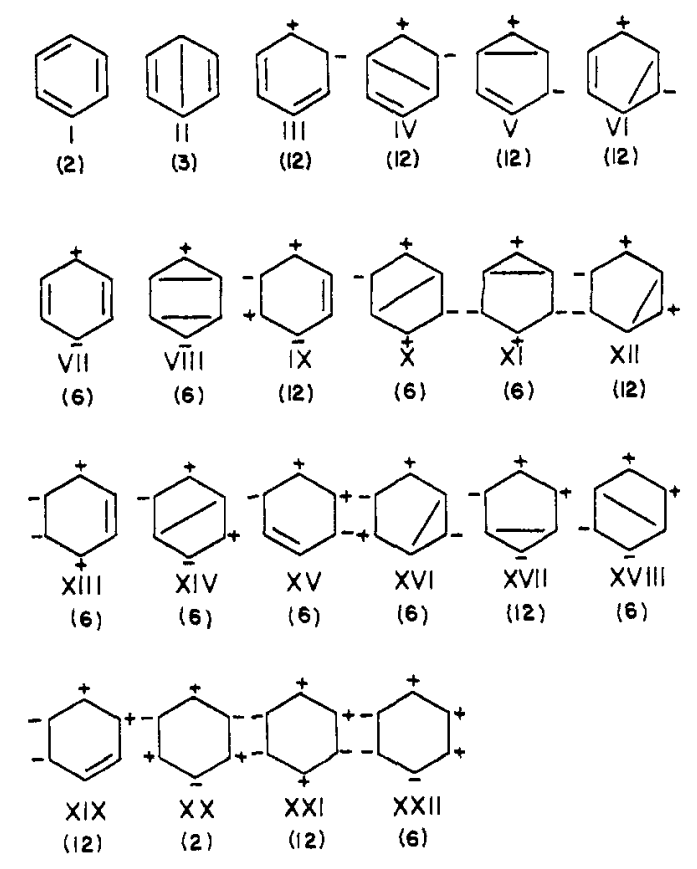
该图取自文献Valence-bond calculation of the electronic structure of benzene
$frag中2*6为C的pz轨道,其中包含了d极化函数,添加了与C相邻的H,使得轨道中可以混入部分H的pz极化函数
一个轨道分配一个片段,添加到$orb末尾(2,3,4,5,6,7),这最后的6个轨道将作为6个活性轨道
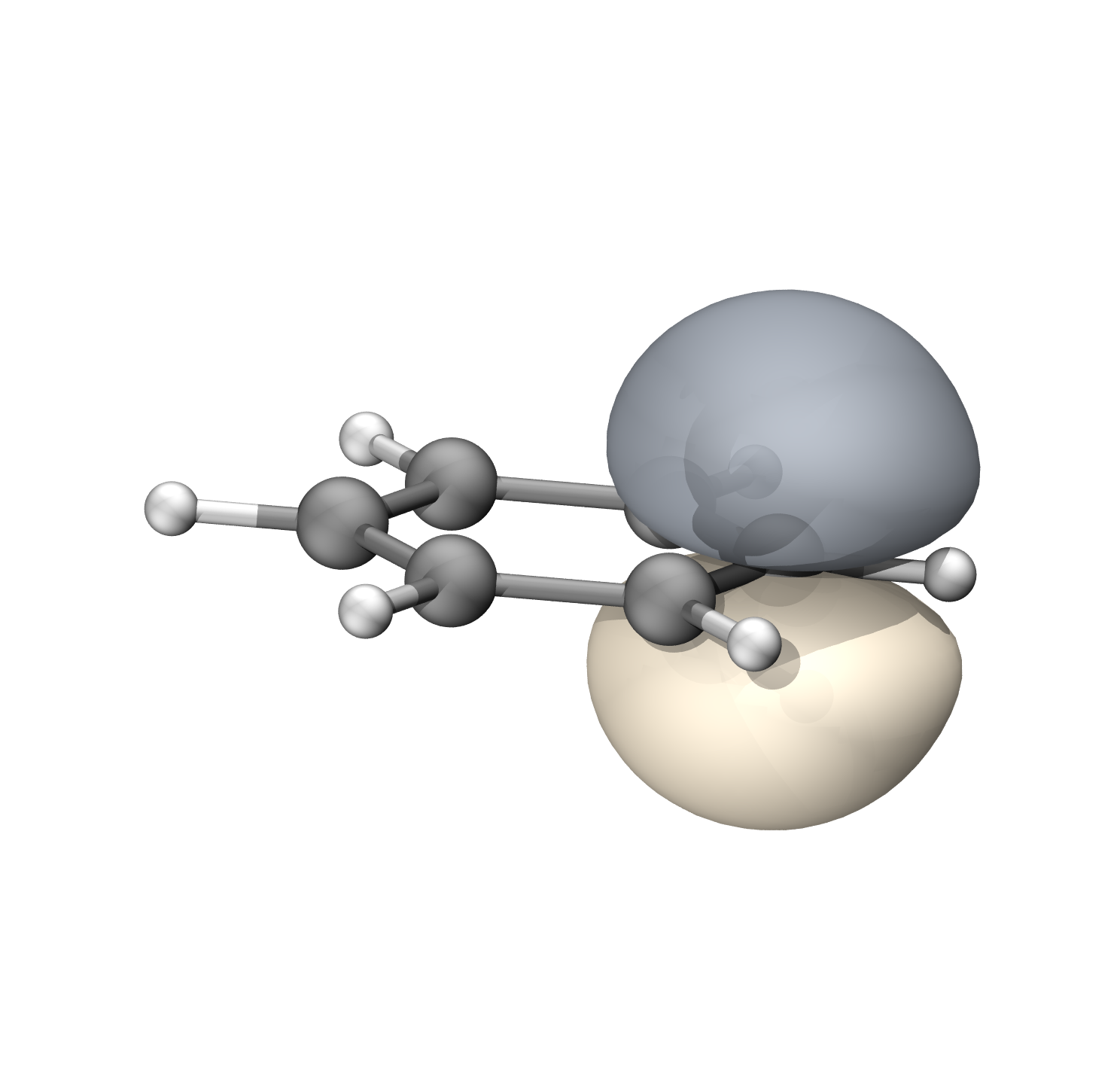
非活性轨道部分为双占据,包含σ部分和C的1s电子,为了简便以及能量最低,使用离域轨道
$frag中定义了一个包含所有原子,除去pzdxzdyz外所有基函数的片段12。并且在$orb中添加了18个包含该片段的轨道,guess=auto将会给出离域分子轨道,填充36个电子,剩余6个π电子作为活性电子
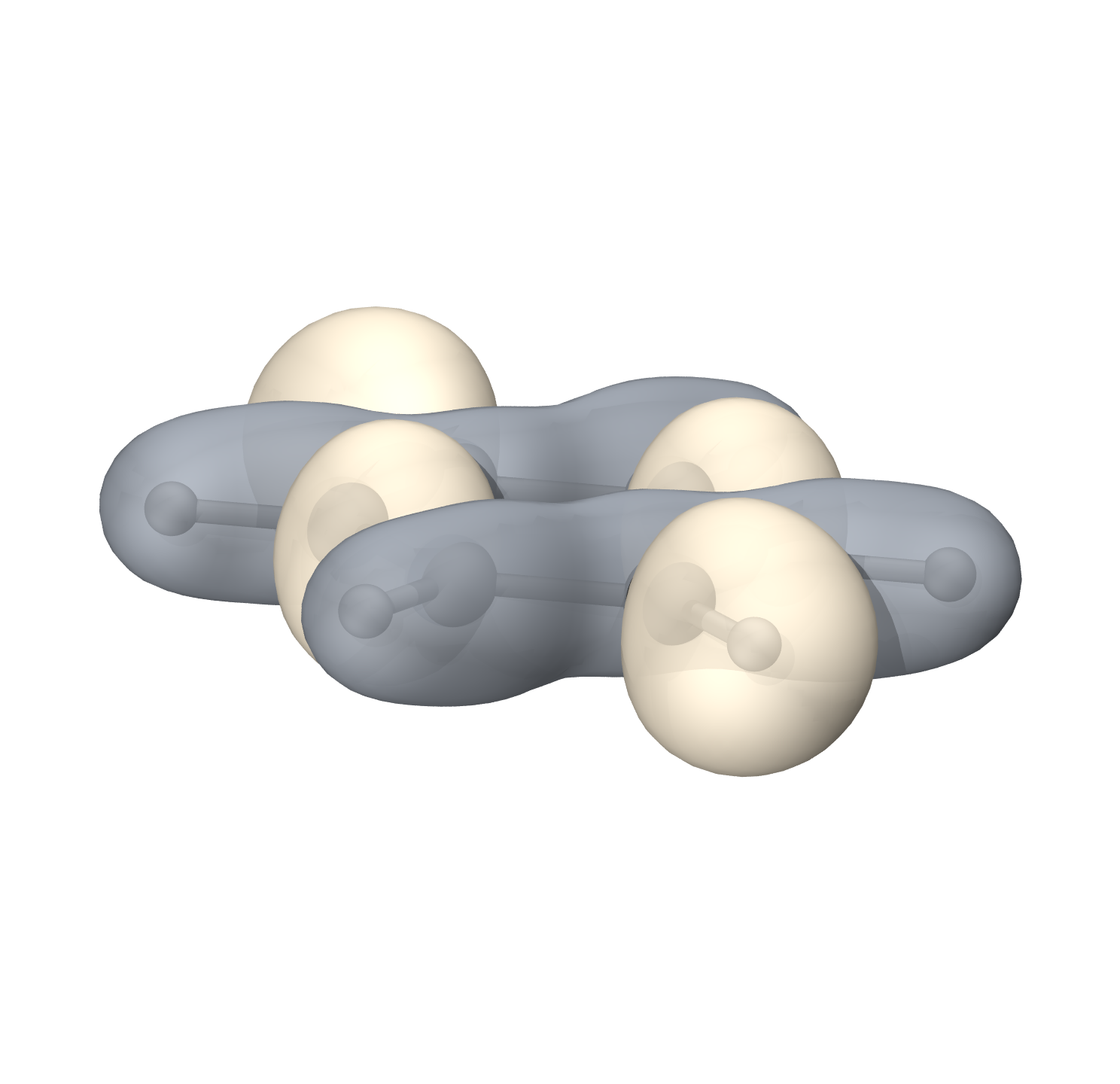 离域轨道其中之一
离域轨道其中之一
仅两种共价结构
将str=full删除,添加nstr=2,在$str中定义共价结构
C6H6
$ctrl
nstr=2 nao=6 nae=6 iscf=5 iprint=3
orbtyp=hao frgtyp=sao
int=libcint basis=cc-pvdz
$end
$frag
12 2*6
spxpydxxdyydzzdxy 1-12
pzdxzdyz 1 2
pzdxzdyz 3 4
pzdxzdyz 5 6
pzdxzdyz 7 8
pzdxzdyz 9 10
pzdxzdyz 11 12
$end
$orb
1*18 1*6
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
2
3
4
5
6
7
$end
$str
1:18 19 20 21 22 23 24
1:18 24 19 20 21 22 23
%end
$geo
C 0.6995000584 1.2115696411 0.0000000000
H 1.2460106991 2.1581538376 0.0000000000
C -0.6995000584 1.2115696411 0.0000000000
H -1.2460106991 2.1581538376 0.0000000000
C -1.3990001169 0.0000000000 0.0000000000
H -2.4920213982 0.0000000000 0.0000000000
C -0.6995000584 -1.2115696411 0.0000000000
H -1.2460106991 -2.1581538376 0.0000000000
C 0.6995000584 -1.2115696411 0.0000000000
H 1.2460106991 -2.1581538376 0.0000000000
C 1.3990001169 0.0000000000 0.0000000000
H 2.4920213982 0.0000000000 0.0000000000
$endstr中两种结构写为:
1:18 19 20 21 22 23 24
1:18 24 19 20 21 22 23前18个轨道为离域的双占据轨道,19-24为pz轨道
5种共价结构
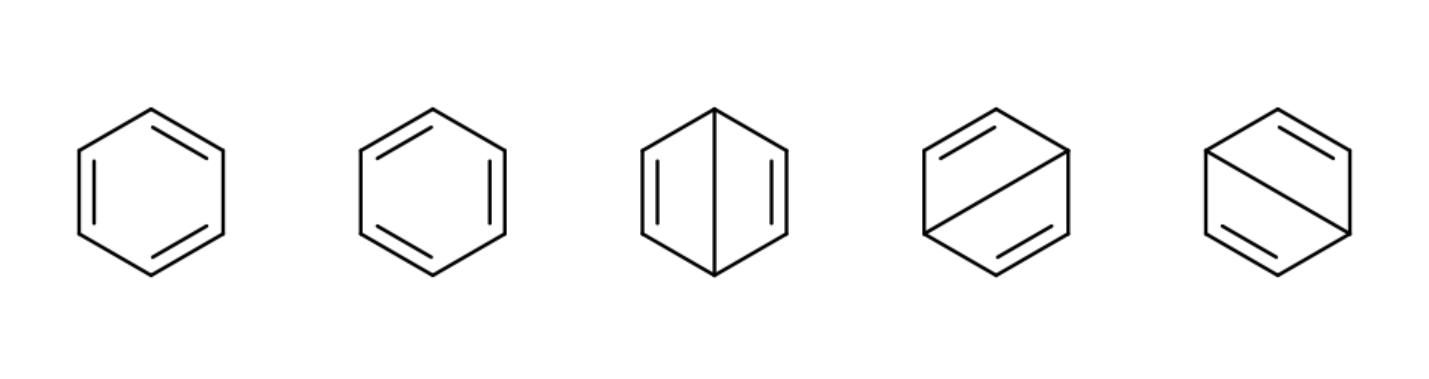
将str=full改为str=cov即可自动生成所有共价结构
str=ion(n,m)可以自动生成所有n阶和m阶离子结构(0阶离子结构为共价结构)
str=ion(n-m)生成所有n阶到m阶离子结构
例如str=ion(0,2,4-6)生成所有0阶、2阶、4阶到6阶离子结构
结果
能量:
| 结构 | 能量(Hartree) |
|---|---|
| 1共价结构 | -230.59097 |
| 2共价结构 | -230.62995 |
| 5共价结构 | -230.63449 |
| full | -230.77728 |
相对于1共价结构的能量:
| 结构 | 能量(kcal/mol) |
|---|---|
| 1共价结构 | 0 |
| 2共价结构 | -24.45995 |
| 5共价结构 | -27.30880 |
| full | -116.90952 |
str=full时结构占比:
| 结构 | Coulson Chirgwin Weight |
|---|---|
| 共价结构I×2 | 21.91% |
| 共价结构II×3 | 10.78% |
| 1阶离子结构III*12 | 24.68% |
| 结构 | Renormalized Weight |
|---|---|
| 共价结构I×2 | 39.83% |
| 共价结构II×3 | 7.57% |
| 1阶离子结构III*12 | 25.69% |
可以看出离子结构是非常重要的MO中50%的离子项
对比一下CASSCF结果:
CASSCF(6,6)能量: -230.79382
| 结构 | 占比 |
|---|---|
| 222000 | 88.46% |
| 211110 | 03.18% |
| 220200 | 02.18% |
| 202020 | 02.18% |
| 121101 | 01.29% |
| 112011 | 01.29% |
占据数:
| A2u | E1g | E1g | E2u | E2u | B2g |
|---|---|---|---|---|---|
| 1.9604 | 1.9004 | 1.9004 | 0.1009 | 0.1009 | 0.0369 |
将6个π轨道定域化再进行CASSCF可以得到相同的能量
| 结构 | 占比 |
|---|---|
| 111111 | 23.98% |
| 1111+2+0 ×12 | 37.32% |
111111对应5个CSF
1111+2+0对应2个CSF
使用软件:
可视化轨道: VMD vcube
CASSCF计算: ORCA 6
VB计算: XMVB
转换XMVB轨道文件 XMVB-tools
计算轨道cube文件: Multiwfn